请问各位老师能发几个[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]采用标准假如法定量的实验例子吗 谢谢
[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法中标准加入法的局限性。回答最全面的有奖![em0815] [em0815] [em0815]
有色谱定量分析中,有标准加入法(standard addition method),标准的加入量是如何确定的呢?
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]器验证中标准溶液哪里有卖
[b][color=#DC143C]【序号】:1【作者】:J.Timm Horst Diehl 杨奇敏 【题名】:[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法中标准加入法的必要条件和限制范围【期刊】:《分析试验室》【年、卷、期、起止页码】: 1984年06期[/color][color=#DC143C]【全文链接】:[url=https://pubs.acs.org/doi/10.1021/ja00462a025]http://www.cnki.com.cn/Article/CJFDTotal-FXSY198406016.htm[/url][/color][color=#DC143C][/color][/b]
[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法是一种相对测量法,必须采用校准的方法来获得未知样品中待测元素的浓度。校准方法是否准确,取决于待测元素在分析样品和校准溶液中是否具有完全相同的分析行为。一旦由于样品中的共存物影响了待测元素的分析行为,使之不同与校准溶液中该元素的行为,则可能使完全相同浓度的溶液给出不同的吸收值,引起干扰。如果对干扰不够重视,未采取相应的消除措施,往往使测定结果不准确。在[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]分析中,常采用标准加入法来抵消干扰,减少分析误差。然而,如果对标准加入法应用不慎,将会引起严重的分析误差,本文将对该法的局限性作一探讨。1、标准加入法的基本原理图1 标准加入法的校准曲线校准加入法[1]是将不同量的标准溶液分别加入数份等体积的试样溶液之中,其中一份试样溶液不加标准,均稀释至相同体积后测定(并制备一个样品空白)。以测定溶液中外加标准物质的浓度为横坐标,以吸光度为纵坐标对应作图,然后将直线延长使之与浓度轴相交,交点对应的浓度值即为试样溶液中待测元素的浓度。标准加入法的曲线如图1所示。图x的绝对值即为测定溶液中被测元素的浓度。在[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分析时,用标准加入法一般须满足三个条件[2]:第一,待测元素浓度从零至最大加入标准浓度范围,必须与吸光度值具有线性关系,并且标准曲线通过坐标原点。第二,在测定溶液中的干扰物质浓度必须恒定。第三,加入标准物质产生的响应值与原样品中待测元素产生的响应值相同。2、标准加入法的存在的一些问题 2.1.浓度的估计和测定的浓度范围在标准加入法中,为获得准确的结果和较好的精密度[2],要求加入标准的浓度系列为样品中待测元素的一倍到数倍。为了确定往样品中加入标准的浓度,就必须估计样品中待测元素的浓度,这使得该操作难以自动化。例如,茶叶消化样中铅的浓度范围为0.005~0.08mg/ml,如果用一种固定的加入量来对待不同的茶叶消化样容易导致结果误差大。响应值与浓度间的线形关系是标准加入法能够成立的基础。在标准加入法中,要求加入标准的浓度系列为样品中待测元素的一倍到数倍,所以可分析的最高浓度只有标准曲线法的1/2~1/5。有研究表明[3],假定[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法的线形范围为0~200个单位,如果要求标准加入法的测定相对标准偏差不得超过5%,那么可用的分析范围为5~40个单位。这比标准曲线法的测定范围窄多了。减少加入量可以扩大测定样品的线形范围,但要以降低精密度为代价。2.2测定的精密度和分析速度由于标准加入法一般测定浓度较低的样品,受方法本身所固有的随机误差影响较大,导致测定的精密度下降。有研究表明[3]:标准加入法在理想条件下所产生结果的标准偏差总比标准曲线法大将近一倍。如果加入标准的浓度不合适,则标准偏差将更加不理想。在标准加入法中,为获得准确的结果和较好的精密度,分析每一个样品都必须进行2~3次加标且加标的浓度应该不同,使得分析速度大大降低。这在进行大规模样品分析中难以推广。在实际样品分析中,有人采用单点的标准加入法,可以使分析速度大大提高,但必须以牺牲精密度和准确度为代价[4]。2.3加和性干扰如果测得的吸光度A中,除了待测元素的吸收B外,还有一个附加的吸收C,且C不随待测元素的浓度而变化(C值可以是正的,也可以是负的),则这种干扰为加和性干扰。加和性干扰的特点是不改变曲线的斜率和形状,只改变曲线的在吸收轴上的截距。光谱线干扰、背景吸收和污染等一般可归于加和性干扰。加和性干扰采用标准加入法是消除不了的。测得的吸光度A=B+C,无论如何加入已知浓度的待测元素,C值是始终消除不了的。校正光谱线干扰和背景吸收的方法是配制尽可能与样品相同基体的标准系列,同时进行背景校正。校正污染需使用完全与样品一样,经过相同前处理的空白作参比。2.4特效性干扰在标准加入法中,加入元素和待测元素从表面上看是同一元素,两者又同处于相同的环境,似乎应该具有完全相同的分析行为。但是,问题并非如此简单,在一定的条件下,即使不同的物种、不同的化合物也可能有不同的分析行为,常常表现为不同浓度的分析元素受到的干扰程度不同,这称为特效性干扰。在火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中,全部的电离干扰和部分的化学干扰都属于特效性干扰,不能通过标准加入法来消除。电离干扰可加入消电离剂(电离电位低元素)等方法来抑制,而特效性的化学干扰可加入稀放剂、保护剂等方法来抑制。在石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中,许多基体干扰(多数的蒸发干扰和全部[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]干扰)是特效性的,不能通过标准加入法来校正。例如,如果标准溶液中的待测元素是以不挥发的无机盐类的形式存在,而样品中的待测元素是以较挥发的有机化合物的形式存在,那么很可能在挥发阶段阶段样品中的待测元素挥发损失了一部分,而标准中的待测元素留下了,显然结果不可能正确。这部分的干扰应通过异构重整或基体改进技术加以抑制。3、运用标准加入法导致错误的实例3.1.火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中特效性的化学干扰[5]。图2 待测元素和样品的浓度变化对分析结果的影响图3钙对铅测定的影响A/A0:相对吸光度;A0:Pb的吸光度;A:有钙干扰时Pb的吸光度(1)如图2中曲线A、B所示,曲线B含有1mg/L钙和12.5mg/L磷,测定其钙的含量结果为1.98mg/L,回收率为198%,而曲线A为2.5mg/L钙和12.5mg/L磷,测定其钙的含量结果为3.49mg/L,回收率为123%。这表明相同含量的干扰介质对不同含量的待测元素产生的干扰效应不同。(2)如图2中曲线A、C所示,而曲线A为2.5mg/L钙和12.5mg/L磷,测定其钙的含量结果为3.49mg/L,回收率为123%。将样品溶液稀释一倍,结果如曲线C所示,钙的含量为2.12 mg/L,换算成原始浓度,结果为4.24 mg/L,回收率为170%,两者相差很大。这表明不同的样品浓度对分析结果的影响不同。测定钙时磷的化学干扰不能通过标准加入法校正,而应加入化学抑制剂(如镧)加以校正。3.2石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中的特效性基体干扰[6]从图3可以看出,同样浓度的钙对铅的干扰程度大小随铅浓度大小的不同而不同,铅的浓度越低吸光度下降越快,随着铅浓度的提高,吸光度下降逐渐减少。这是石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中典型的蒸发和[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]干扰,是属于与浓度无关的特效性干扰。因此,当用标准加入法测定铅时,这种干扰不仅影响校准曲线的斜率,而且会使校准曲线发生弯曲,对一同样浓度的钙,铅的浓度越小,校准曲线向纵轴弯曲。因此,但存在与浓度有关的特效性干扰时,不能通过校准加入法来校正,而必须采用合适的手段(如加基体改进剂)来克服干扰,才能得到准确的结果。4、结论(1)与标准曲线法相比,校准加入法测定的浓度范围变窄,精密度下降,操作烦琐,分析效率大大降低;(2)校准加入法不能消除加和性干扰,如光谱线干扰,背景吸收和污染等;(3)校准加入法不能消除特效性干扰,如火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中电离干扰和化学干扰,石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中的特效性基体干扰。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=45413][url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法中标准加入法的局限性[/url]
最近碰到两个不可分开的组分,实验室条件下,实在不能找到其他办法,于是我想到标准加入法,因为其中一个组分是有标准品的,这个方法[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]有效,是否有人用在GC上呢?欢迎讨论!
原子吸收光谱法是一种相对测量法,必须采用校准的方法来获得未知样品中待测元素的浓度。校准方法是否准确,取决于待测元素在分析样品和校准溶液中是否具有完全相同的分析行为。一旦由于样品中的共存物影响了待测元素的分析行为,使之不同与校准溶液中该元素的行为,则可能使完全相同浓度的溶液给出不同的吸收值,引起干扰。如果对干扰不够重视,未采取相应的消除措施,往往使测定结果不准确。在原子吸收光谱分析中,常采用标准加入法来抵消干扰,减少分析误差。然而,如果对标准加入法应用不慎,将会引起严重的分析误差,本文将对该法的局限性作一探讨。校准加入法是将不同量的标准溶液分别加入数份等体积的试样溶液之中,其中一份试样溶液不加标准,均稀释至相同体积后测定(并制备一个样品空白)。以测定溶液中外加标准物质的浓度为横坐标,以吸光度为纵坐标对应作图,然后将直线延长使之与浓度轴相交,交点对应的浓度值即为试样溶液中待测元素的浓度。标准加入法的曲线如图1所示。图x的绝对值即为测定溶液中被测元素的浓度。在原子吸收分析时,用标准加入法一般须满足三个条件:第一,待测元素浓度从零至最大加入标准浓度范围,必须与吸光度值具有线性关系,并且标准曲线通过坐标原点。第二,在测定溶液中的干扰物质浓度必须恒定。第三,加入标准物质产生的响应值与原样品中待测元素产生的响应值相同。2、标准加入法的存在的一些问题2.1.浓度的估计和测定的浓度范围在标准加入法中,为获得准确的结果和较好的精密度,要求加入标准的浓度系列为样品中待测元素的一倍到数倍。为了确定往样品中加入标准的浓度,就必须估计样品中待测元素的浓度,这使得该操作难以自动化。例如,茶叶消化样中铅的浓度范围为0.005~0.08mg/ml,如果用一种固定的加入量来对待不同的茶叶消化样容易导致结果误差大。响应值与浓度间的线形关系是标准加入法能够成立的基础。在标准加入法中,要求加入标准的浓度系列为样品中待测元素的一倍到数倍,所以可分析的最高浓度只有标准曲线法的1/2~1/5。有研究表明[,假定原子吸收法的线形范围为0~200个单位,如果要求标准加入法的测定相对标准偏差不得超过5%,那么可用的分析范围为5~40个单位。这比标准曲线法的测定范围窄多了。减少加入量可以扩大测定样品的线形范围,但要以降低精密度为代价。2.2测定的精密度和分析速度由于标准加入法一般测定浓度较低的样品,受方法本身所固有的随机误差影响较大,导致测定的精密度下降。有研究表明:标准加入法在理想条件下所产生结果的标准偏差总比标准曲线法大将近一倍。如果加入标准的浓度不合适,则标准偏差将更加不理想。在标准加入法中,为获得准确的结果和较好的精密度,分析每一个样品都必须进行2~3次加标且加标的浓度应该不同,使得分析速度大大降低。这在进行大规模样品分析中难以推广。在实际样品分析中,有人采用单点的标准加入法,可以使分析速度大大提高,但必须以牺牲精密度和准确度为代价。2.3加和性干扰如果测得的吸光度A中,除了待测元素的吸收B外,还有一个附加的吸收C,且C不随待测元素的浓度而变化(C值可以是正的,也可以是负的),则这种干扰为加和性干扰。加和性干扰的特点是不改变曲线的斜率和形状,只改变曲线的在吸收轴上的截距。光谱线干扰、背景吸收和污染等一般可归于加和性干扰。加和性干扰采用标准加入法是消除不了的。测得的吸光度A=B+C,无论如何加入已知浓度的待测元素,C值是始终消除不了的。校正光谱线干扰和背景吸收的方法是配制尽可能与样品相同基体的标准系列,同时进行背景校正。校正污染需使用完全与样品一样,经过相同前处理的空白作参比。2.4特效性干扰在标准加入法中,加入元素和待测元素从表面上看是同一元素,两者又同处于相同的环境,似乎应该具有完全相同的分析行为。但是,问题并非如此简单,在一定的条件下,即使不同的物种、不同的化合物也可能有不同的分析行为,常常表现为不同浓度的分析元素受到的干扰程度不同,这称为特效性干扰。在火焰原子吸收中,全部的电离干扰和部分的化学干扰都属于特效性干扰,不能通过标准加入法来消除。电离干扰可加入消电离剂(电离电位低元素)等方法来抑制,而特效性的化学干扰可加入稀放剂、保护剂等方法来抑制。在石墨炉原子吸收中,许多基体干扰(多数的蒸发干扰和全部气相干扰)是特效性的,不能通过标准加入法来校正。例如,如果标准溶液中的待测元素是以不挥发的无机盐类的形式存在,而样品中的待测元素是以较挥发的有机化合物的形式存在,那么很可能在挥发阶段阶段样品中的待测元素挥发损失了一部分,而标准中的待测元素留下了,显然结果不可能正确。这部分的干扰应通过异构重整或基体改进技术加以抑制。3、结论(1)与标准曲线法相比,校准加入法测定的浓度范围变窄,精密度下降,操作烦琐,分析效率大大降低;(2)校准加入法不能消除加和性干扰,如光谱线干扰,背景吸收和污染等;(3)校准加入法不能消除特效性干扰,如火焰原子吸收中电离干扰和化学干扰,石墨炉原子吸收中的特效性基体干扰。
在色谱定量分析中,有标准加入法(standard addition method),标准的加入量是如何确定的呢?
请教各位老师: 在用气相色谱做有机溶剂残留时,质量标准中标准溶液配制方法如下:取无水乙醇约50mg,乙酸乙酯10mg,二氯甲烷60mg,均精密称定,分别置于500ml的容量瓶中,加水溶解并稀释至刻度,摇匀,即可。这里面我有以下几个疑问:1 在试验操作时,称取无水乙醇50mg,置于500ml容量瓶中,由于无水乙醇极易挥发,如果直接称到500ml的容量瓶中的话,已称好的无水乙醇不是会容易挥发吗,有可能实际由50mg挥发到40mg?这样标准品浓度不是降低了吗?误差不是很大?有没有必要先在容量瓶中加入适量的溶剂,再加入挥发性有机溶剂标准品?平时大家做有机溶剂残留,有机溶剂大多易挥发,如何保证已称好的标准品不挥发损失呢?2 气相有机溶剂残留有机溶剂标准品用分析纯的可以吗?如果分析纯纯度是大于99%,在计算标准品浓度时是按99%还是100%纯度计算?另外分析纯含有的杂质产生的杂质峰怎么办?3 气相配制标准品或样品溶液时,一般用什么溶剂来配制,是只要对标准品与样品溶解性好的溶剂吗?为什么经常有人用DMF(NN-二甲基甲酰胺),说是万能溶剂,也有用DMSO或水的,一般如何选择?
请教一下各位,气相色谱法中的标准加入法的公式是什么?
色谱测定中标准曲线取几个点合适
大家好!我和同事要做稳定性试验了,现在还在写文件阶段,欧洲药典要求[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法用标准加入法。哪位同仁可以介绍一下标准加入法怎么测残留溶剂含量啊。最好有实例。谢谢了!
液相色谱分析中标准样的浓度一般是多少?我是混合标准样
上海安谱实验科技股份有限公司参加由上海市机械设备成套(集团)有限公司组织的上海疾病预防控制中心甲状腺项目包和标准品及色谱耗材招标,于2017年12月,成功中标4个包件,包括甲状腺项目包1、甲状腺项目包3、甲状腺项目包6、标准品及色谱耗材,中标总金额达104.7万。 甲状腺项目包1中标公告: [align=center][img]http://www.anpel.com.cn/UpFile/Admin/image/20180111/20180111165812_7829.png[/img] [/align] 甲状腺项目包3中标公告: [align=center][img]http://www.anpel.com.cn/UpFile/Admin/image/20180111/20180111165851_9260.png[/img] [/align] 甲状腺项目包6中标公告: [align=center][img]http://www.anpel.com.cn/UpFile/Admin/image/20180111/20180111165917_6613.png[/img] [/align] 标准品及色谱耗材中标公告: [align=center][img]http://www.anpel.com.cn/UpFile/Admin/image/20180111/20180111165944_4002.png[/img] [/align]
请问顶空[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定药物有机溶剂残留量,1、如何提高分析灵敏度?2、定量方法选外标、内标还是标准加入法为好?
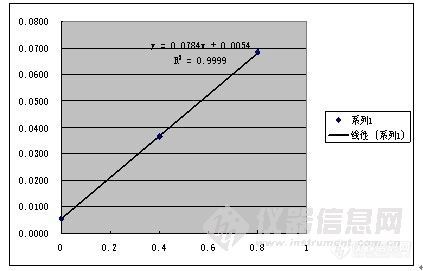
离子色谱标准加入法测凝析水中的硫酸根凝析水是凝析油经油气分离后液相析出的水。其特点是矿化度比较低,pH值偏酸性。各离子含量不高,一般用标准曲线法可以进行定量测定。但是最近遇到几个水样的结果有点奇怪。在此把本人的分析汇报一下,看思路和作法有何不妥希望专家给予指导。一、 奇怪的现象:水样分析发现稀释后测定结果比原液测定结果高,而且不同的稀释倍数得出不同测定结果。再次重新绘制标准曲线,并测定原液和不同稀释倍数的样品,依然如故。结果如下http://ng1.17img.cn/bbsfiles/images/2011/08/201108211902_311443_1608710_3.jpg二、 原因分析针对此种情况,初步分析原因如下:1由于测定结果均于标准线性的下限处,导致结果的不确定性。2.原样品存在一定的基体干扰于是,第二步,用标准加入法测定原液和不同稀释倍数的样品,结果如下:http://ng1.17img.cn/bbsfiles/images/2011/08/201108211903_311444_1608710_3.jpg其中,S1=0.4mg/L,S2=0.8 mg/L.上述结果分别对应下面三个图:http://ng1.17img.cn/bbsfiles/images/2011/08/201108211903_311445_1608710_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/08/201108211904_311446_1608710_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/08/201108211904_311447_1608710_3.jpg以上三个结果的斜率均与标准曲线斜率0.2047相差较大,说明可能的确存在基体干扰。另外可以看出,稀释倍数越大,所做标准加入法的线性也越好。三个结果的线性以最后一个线性最好达到1.0000。 而且这个结果与最终的离子和计算相吻合。因此最后以此为最终结果报出。三、 我的疑惑1. 按照DL=2N/S的方法,用离子色谱60分钟的基线噪声为0.0077,则DL=0.075 mg/L。(是不是有点高了,印象中离子色谱的DL很低的呀)而所测结果0.414 mg/L、0.28 mg/L、0.22 mg/L,均高于0.075 mg/L很多,所测结果应该可靠的。可是又与标准加入法的结果相差很大。如何解释?如果说的确有基体干扰存在,而水样本身总离子量比较小,又经过RP柱和Ag/H柱过滤后的,基体有什么能干扰呢?2. 从标准加入法结果看,稀释100倍后线性正相关性很高。可是标准加入的量S1=0.8 mg/L,S2=0.4mg/L,而测得结果却只有0.0685 mg/L,似乎标准加入的量是不合适的;稀释20倍后线性正相关性达到3个9。而测得结果却只有0.108 mg/L,似乎标准加入的量也是不合适的;不稀释的原液标准加入的线性正相关性较差,只有1个9。但测得结果0.637 mg/L,却处于S1=0.8 mg/L和S2=0.4mg/L之间,标准加入的量应是合适的。而且此结果与标准曲线法的原液测定结果相近。如果它是真值,又如何解释稀释倍数越大,测定结果也越大呢?

GB/T 23768-2009 无机化工产品 火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法通则 中应用标准加入法时要求彻底校正背景,怎么确定是否彻底校正背景?[img=,690,440]https://ng1.17img.cn/bbsfiles/images/2019/01/201901011020508705_4385_1638724_3.jpg!w690x440.jpg[/img]
GC4002气象色谱2010工作站中标准曲线怎么做?做油里的6#溶剂的残溶,曲线怎么做的,具体一点,参数设置什么的都要,很急,拜托大家了!
近日,根据财政部预算安排要求,中国环境监测总站为国家环境监测网地表水饮用水源地水环境质量监测集中采购一批标准样品,要求标准品必须满足可用于《地表水环境质量标准》(GB3838-2002)表3中地表水环境质量监测中包括挥发性有机物类,半挥发性有机污染物类,有机磷农药类,无机元素类等80项特定指标的测试;此次招标要求投标方必须是在中华人民共和国境内注册、注册资金至少在500万元人民币以上、具有独立法人资格的单位,必须通过ISO9000质量管理体系认证,并有良好的商业信誉和所投产品业绩等。迪马科技凭借优良的产品特性和品质,极高的性价比以及完善的服务,在众多竞争对手中脱颖而出,成功中标中国环境监测总站地表水环境质量监测标准样品采购项目,中标金额为194.9995万元。中标详情如下:http://www.dikma.com.cn/Public/Uploads/images/1(5).jpg迪马科技自成立将近二十年来,一直致力于研发制造科学、高效的化学分析产品,提供完善服务和全面解决方案,在色谱填料研发,色谱柱制造和相关分离产品等多个技术领域始终保持世界先进水平。ProElut 固相萃取小柱,DikmaCap系列气相色谱柱,Diamonsil钻石二代通用型反相色谱柱、1.8μm Endeavorsil UHPLC超高压液相色谱柱、2.7μm Leapsil HPLC/UHPLC兼容色谱柱、Bio-Bond 300 Å蛋白和多肽专用色谱柱、Inspire高性能宽pH反相色谱柱、Spursil极性改性色谱柱等六大液相色谱柱在其不断的实际应用中,展现了其出色的性能。以下为我司中标的部分标准品:注:1. 以下为有现货的部分标准品,更多地表水检测化学标准品,欢迎来电垂询;2. 表中未列出的三氯甲烷、丙烯腈、对硫磷、敌敌畏、黄磷等多数为管制产品;3. 表中“编号”系指与《地表水环境质量标准》(GB3838-2002)表3相对应的编号
标准溶液配制过程中标准物质如何移取今天群里有友友问如果用标准物质配制标准溶液,用移液枪,移液管还是什么移取更准确些呢?http://simg.instrument.com.cn/bbs/images/default/emyc1010.gifhttp://simg.instrument.com.cn/bbs/images/default/em09511.gif好多实验猿们都应该遇到过这种情况吧!?http://simg.instrument.com.cn/bbs/images/default/emyc1010.gif平时标准物质都是装在1ml的安瓿瓶,打开瓶子,如何吸取呢?http://simg.instrument.com.cn/bbs/images/default/em09507.gif针对这个问题群友展开了讨论,你觉得什么更准,请发表见解继续回复帖子!http://simg.instrument.com.cn/bbs/images/default/em09511.gif
麻烦懂行的达达们帮我设计个气相色谱外标法标准曲线的操作流程。标准样品有4针3个不同浓度,一针是空白。请问标准样品需要稀释到一定浓度还是可以直接进样。还有怎么做标准曲线啊?
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]内标法和外标法作标准曲线有何不同?
有谁用过[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]质谱联用仪定量模块的标准加入法定量(Std Addition),具体怎么用?
[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法是一种相对测量法,必须采用校准的方法来获得未知样品中待测元素的浓度。校准方法是否准确,取决于待测元素在分析样品和校准溶液中是否具有完全相同的分析行为。一旦由于样品中的共存物影响了待测元素的分析行为,使之不同与校准溶液中该元素的行为,则可能使完全相同浓度的溶液给出不同的吸收值,引起干扰。如果对干扰不够重视,未采取相应的消除措施,往往使测定结果不准确。在[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]分析中,常采用标准加入法来抵消干扰,减少分析误差。然而,如果对标准加入法应用不慎,将会引起严重的分析误差,本文将对该法的局限性作一探讨。1、标准加入法的基本原理 校准加入法是将不同量的标准溶液分别加入数份等体积的试样溶液之中,其中一份试样溶液不加标准,均稀释至相同体积后测定(并制备一个样品空白)。以测定溶液中外加标准物质的浓度为横坐标,以吸光度为纵坐标对应作图,然后将直线延长使之与浓度轴相交,交点对应的浓度值即为试样溶液中待测元素的浓度。在[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分析时,用标准加入法一般须满足三个条件[2]:第一,待测元素浓度从零至最大加入标准浓度范围,必须与吸光度值具有线性关系,并且标准曲线通过坐标原点。第二,在测定溶液中的干扰物质浓度必须恒定。第三,加入标准物质产生的响应值与原样品中待测元素产生的响应值相同。2、标准加入法的存在的一些问题2.1.浓度的估计和测定的浓度范围在标准加入法中,为获得准确的结果和较好的精密度,要求加入标准的浓度系列为样品中待测元素的一倍到数倍。为了确定往样品中加入标准的浓度,就必须估计样品中待测元素的浓度,这使得该操作难以自动化。例如,茶叶消化样中铅的浓度范围为0.005~0.08mg/ml,如果用一种固定的加入量来对待不同的茶叶消化样容易导致结果误差大。响应值与浓度间的线形关系是标准加入法能够成立的基础。在标准加入法中,要求加入标准的浓度系列为样品中待测元素的一倍到数倍,所以可分析的最高浓度只有标准曲线法的1/2~1/5。有研究表明,假定[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法的线形范围为0~200个单位,如果要求标准加入法的测定相对标准偏差不得超过5%,那么可用的分析范围为5~40个单位。这比标准曲线法的测定范围窄多了。减少加入量可以扩大测定样品的线形范围,但要以降低精密度为代价。2.2测定的精密度和分析速度由于标准加入法一般测定浓度较低的样品,受方法本身所固有的随机误差影响较大,导致测定的精密度下降。有研究表明:标准加入法在理想条件下所产生结果的标准偏差总比标准曲线法大将近一倍。如果加入标准的浓度不合适,则标准偏差将更加不理想。在标准加入法中,为获得准确的结果和较好的精密度,分析每一个样品都必须进行2~3次加标且加标的浓度应该不同,使得分析速度大大降低。这在进行大规模样品分析中难以推广。在实际样品分析中,有人采用单点的标准加入法,可以使分析速度大大提高,但必须以牺牲精密度和准确度为代价[4]。2.3加和性干扰如果测得的吸光度A中,除了待测元素的吸收B外,还有一个附加的吸收C,且C不随待测元素的浓度而变化(C值可以是正的,也可以是负的),则这种干扰为加和性干扰。加和性干扰的特点是不改变曲线的斜率和形状,只改变曲线的在吸收轴上的截距。光谱线干扰、背景吸收和污染等一般可归于加和性干扰。加和性干扰采用标准加入法是消除不了的。测得的吸光度A=B+C,无论如何加入已知浓度的待测元素,C值是始终消除不了的。校正光谱线干扰和背景吸收的方法是配制尽可能与样品相同基体的标准系列,同时进行背景校正。校正污染需使用完全与样品一样,经过相同前处理的空白作参比。2.4特效性干扰在标准加入法中,加入元素和待测元素从表面上看是同一元素,两者又同处于相同的环境,似乎应该具有完全相同的分析行为。但是,问题并非如此简单,在一定的条件下,即使不同的物种、不同的化合物也可能有不同的分析行为,常常表现为不同浓度的分析元素受到的干扰程度不同,这称为特效性干扰。在火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中,全部的电离干扰和部分的化学干扰都属于特效性干扰,不能通过标准加入法来消除。电离干扰可加入消电离剂(电离电位低元素)等方法来抑制,而特效性的化学干扰可加入稀放剂、保护剂等方法来抑制。在石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中,许多基体干扰(多数的蒸发干扰和全部[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]干扰)是特效性的,不能通过标准加入法来校正。例如,如果标准溶液中的待测元素是以不挥发的无机盐类的形式存在,而样品中的待测元素是以较挥发的有机化合物的形式存在,那么很可能在挥发阶段阶段样品中的待测元素挥发损失了一部分,而标准中的待测元素留下了,显然结果不可能正确。这部分的干扰应通过异构重整或基体改进技术加以抑制。3、运用标准加入法导致错误的实例3.1.火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中特效性的化学干扰3.2石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中的特效性基体干扰同样浓度的钙对铅的干扰程度大小随铅浓度大小的不同而不同,铅的浓度越低吸光度下降越快,随着铅浓度的提高,吸光度下降逐渐减少。这是石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中典型的蒸发和[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]干扰,是属于与浓度无关的特效性干扰。因此,当用标准加入法测定铅时,这种干扰不仅影响校准曲线的斜率,而且会使校准曲线发生弯曲,对一同样浓度的钙,铅的浓度越小,校准曲线向纵轴弯曲。因此,但存在与浓度有关的特效性干扰时,不能通过校准加入法来校正,而必须采用合适的手段(如加基体改进剂)来克服干扰,才能得到准确的结果。4、结论(1)与标准曲线法相比,校准加入法测定的浓度范围变窄,精密度下降,操作烦琐,分析效率大大降低;(2)校准加入法不能消除加和性干扰,如光谱线干扰,背景吸收和污染等;(3)校准加入法不能消除特效性干扰,如火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中电离干扰和化学干扰,石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]中的特效性基体干扰。
气相色谱定量分析中内标法和标准曲线法有什么区别?感觉差不多,都得绘制标准曲线,这两个曲线有什么区别?
哪位手里有污水处理厂[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法分析项目标准,在下没接触过[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],但现在要开,没找到分析方法,
[font=黑体][color=black]发帖人:[/color][/font]https://bbs.instrument.com.cn/topic/76768029[font=黑体][color=black][b]问题描述:[/b][/color][/font] HJ735-2015[font=宋体]是用来测定土壤和沉积物中的挥发性卤代烃的,采用[/font]sim[font=宋体]法定量。其中标准里把浓度高于[/font]200[font='Cambria Math','serif']μ[/font]g/kg[font=宋体]的样品作为高浓度样品处理,采用甲醇提取稀释的方法测定。低于[/font]200[font='Cambria Math','serif']μ[/font]g/kg[font=宋体]的是低浓度样品,加水直接吹扫进样。可是标准里面给的曲线范围最高点只到[/font]100ng[font=宋体],也即[/font]20[font='Cambria Math','serif']μ[/font]g/kg[font=宋体]。那么[/font]20[font='Cambria Math','serif']μ[/font]g/kg[font=宋体]到[/font]200[font='Cambria Math','serif']μ[/font]g/kg[font=宋体]这个浓度范围的样品怎么定量?是否需要做方法确认?[/font][font=黑体][color=black][b]解答:[/b][/color][/font]1[font=宋体].《土壤和沉积物[/font] [font=宋体]挥发性卤代烃的测定[/font] [font=宋体][url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url][/font]/[font=宋体]质谱法》([/font]HJ735-2015[font=宋体])中定量方式采用[/font]SIM[font=宋体]法,目标物的检出限:[/font]0.3μg/kg[font=宋体],校准曲线范围:[/font]5~100 ng[font=宋体]。[/font][font=宋体]增加高浓度点标准系列,不算方法偏离,不用进行方法确认。但是因为该标准中的检出限很低,过高的浓度点的加入会影响低浓度点定量,同时可能造成实验室检出限达不到标准中的要求。[/font]1.[font=宋体]现场使用便携式[/font]VOCs[font=宋体]检测仪([/font]PID[font=宋体])对样品进行初筛,初步判定样品中挥发性有机物的含量小于[/font]20 μg/kg[font=宋体]时,采集[/font]5g[font=宋体]样品于吹扫瓶内,[/font]20μg/kg-200μg/kg[font=宋体]时,采集[/font]1g[font=宋体]样品于吹扫瓶内。平行采集[/font]3[font=宋体]份,直接分析;大于[/font]200μg/kg[font=宋体]时,采集[/font]5g[font=宋体]样品于预先装有[/font]10 mL[font=宋体]甲醇的吹扫瓶内,平行采集三份。同时采集样品于[/font]60 mL[font=宋体]棕色样品瓶内,并尽量填满,用于样品含水率的测定。[/font]
气相色谱标准加入法测定环己酮中15种微量杂质 环己酮作为一种重要的化工原料和化工溶剂,是制造己内酰胺、己二酸和尼龙的重要中间体。目前环己酮的生产工艺主要有环己烷液相氧化,苯酚加氢、环己烯水合等多种方法,超过90%的环己酮是通过环己烷氧化的工艺生产的。工业用已内酰胺的产量逐年在增加,对环己酮的需求量也越来越大,随着已内酰胺高端市场的需求,对环己酮中杂质含量的控制要求严格。现有的环己酮产品检验方法主要依据工业用环己酮检验标准(GB/T10669-2001),采用填充柱分离,效果差。近年来,有关环己酮产品新的分析方法文献较少,对于环己酮中微量的环己烷、己醛、丁基环己烷、丁醇、4-庚酮、3-庚酮、2-庚酮、戊基环己烷、戊醇、环戊醇、2-甲基环己酮、3-甲基环己酮、4-甲基环己酮、环己醇等微量杂质达不到同时监控的效果,且分析误差较大。 本方法最终使用CP-Wax 52CB50m×0.32mm×1.2µm毛细管柱,程序升降温控制进行分离,采用外标法定量,同时测定环己酮中微量的环己烷、戊醛、己醛、丁基环己烷、丁醇、4-庚酮、3-庚酮、2-庚酮、戊基环己烷、戊醇、环戊醇、2-甲基环己酮、3-甲基环己酮、4-甲基环己酮、环己醇等15种杂质,分离度均大于1.5,且准确度高。1 实验部分1.1试剂与仪器 环己酮为南化公司生产,环己烷、戊醛、己醛、丁基环己烷、丁醇、4-庚酮、3-庚酮、2-庚酮、戊基环己烷、戊醇、环戊醇、2-甲基环己酮、3-甲基环己酮、4-甲基环己酮、环己醇为色谱纯。 Agilent7890气相色谱仪,FID检测器,Agilent Chemstaion色谱工作站,毛细管色谱柱 CP-Wax 52 CB 50m×0.32mm×1.2µm,1µl微量进样针。1.2 实验条件 汽化室温度250℃,检测器温度300℃,初始柱温115℃,程序升降温控制进行分离,柱流量0.8ml/min(恒流),分流比30:1,高纯He为载气,进样量为0.4µl。1.3[/f
急!急!急!麻烦懂行的达达们帮我设计个气相色谱外标法标准曲线的操作流程。是做板材中TVOC的含量的,标准样品有4针3个不同浓度,一针是空白。请问标准样品需要稀释到一定浓度还是可以直接进样。还有怎么做标准曲线啊?







 我要推广仪器
我要推广仪器
 下载APP
下载APP