液相色谱串联质谱法的原理
近日,中国国家标准化管理委员会(以下简称“国标委”)发布了《液相色谱-串联四极质谱仪性能的测定方法》(GB/T 35410-2017)。该国家标准收录在2017年第32号中国国家标准公告中,将于2018年4月1日开始实施。该标准由国家科技部提出,由全国仪器分析测试标准化技术委员会归口,起草单位是中国计量科学研究院。该国家标准规定了液相色谱-串联四极质谱仪性能的测试方法,适用于液相色谱-串联四极质谱仪性能的测定。 液相色谱-串联四极质谱仪作为最有代表性的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]类型,广泛应用于食品、药品、环境、化工、临床、科研等领域,几乎覆盖了国计民生的方方面面。2017年,我国采购的液相色谱-串联四极质谱仪总量超过1000台,总金额约在15亿元到20亿元之间。目前,我国尚不具备成熟的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用仪[/color][/url]生产能力,主要靠进口。目前市场上液相色谱-串联四极质谱仪的主流品牌多达6-7家,型号更是繁多,普通购买者没有办法快速、直观地了解每台仪器的性能。该国家标准的出台,树立了统一的仪器性能评价标准,有助于对不同品牌、型号的仪器参比和行业秩序的改善。也有助于产品研发时做技术评价。 以下为详细内容:[url]http://www.instrument.com.cn/news/20180126/238974.shtml[/url]
高效液相色谱串联质谱可以定量吗?
刚接触液相色谱-串联质谱,请问用液相色谱-串联质谱时,为什么要使用两种模式(正离子电喷雾、负离子电喷雾)才能对物质进行定性呢?还有两种模式(正离子电喷雾、负离子电喷雾)的区别是什么时,如SN/T1948-2007甜蜜素的负离子电喷雾,定性、定量的离子对为178。2/79。8;而在正离子电喷雾下,则定性、定量的离子对却为202。2/122。2啊,新手,多谢各位的帮助
超高效液相色谱串联质谱法原理
高效液相色谱串联质谱法分析烟草中15中农药残留烟草化学 2011年第7期
有版友询问,谁有waters液相色谱串联质谱软件的操作说明呢,可否分享一下?ACQUITY TM UPLC,TQMS
[b]超高效液相色谱串联质谱法是什么方法[/b]
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]串联质谱仪原理是什么
序号:作者:倪姮佳; 黄显会; 方炳虎; 贺利民; 赵永达;期刊:分析测试学报,题名:高效液相色谱-串联质谱法测定猪组织中的土拉霉素年号:2011年03期链接:http://202.119.208.220:8002/kns50/detail.aspx?dbname=CJFDTEMP&filename=TEST201103019
我用液相串联二级质谱测一组混合物中的物质,一级质谱的质核比结果只能对应到小数点后一位,二级质谱打碎后的粒子质核比在文献中没有找到参考数据,这该怎么办呢?我可以通过跑标品的液相色谱确定该物质存在吗?
请问各位老师,Waters[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]串联质谱仪有Xevo TQ-XS、Xevo TQ-S、Xevo TQ-S micro等几个型号,求教这三个型号的差异,谢谢
液相色谱串联质谱用的标准品可以去哪里购买?
[b]高效液相色谱三重四极杆串联质谱联用仪可以进行哪些工作模式?以及采用这些工作模式有什么作用?[/b]
哪位老师提供一个液相色谱串联质谱的检验报告的模板啊,就是检测人员填的样品名称,温度湿度,色谱条件,最后结果等等我的信箱: wittyzhou@163.com谢谢了!
【序号】:1【作者】:沈洲; 沈鸿烈; 马跃; 陈军; 【题名】:液相色谱串联质谱法测定牛组织中氨丙啉残留量【期刊】:分析化学 【链接】:http://www.cnki.net/kcms/detail/detail.aspx?QueryID=14&CurRec=120&DbCode=CJSF&dbname=CJSFLAST2011&filename=BDTQ201102006
[color=#444444]请问用液相色谱串联质谱法测试水样中农药含量时,为什么要在过滤后的水样中加入等体积的有机溶剂?最终如何计算测得的浓度?是否为2倍关系?[/color]
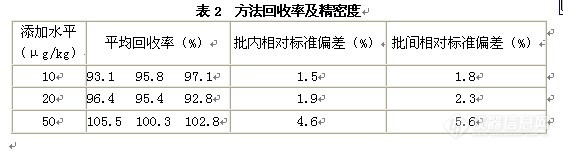
高效液相色谱串联质谱测定猪肉中新型兽药泰拉霉素摘要 建立了猪肉中新型兽药泰拉霉素的液相色谱串联质谱(HPLC-MS/MS)联用确证方法。样品用甲醇+0.1%磷酸(70:30 v/v)提取,离心后用PCX固相萃取小柱净化,Symmetry®C8色谱柱分离,串联质谱多反应监测(MRM)模式下分析,内标法定量。方法的线性范围为10~500μg/kg,检出限为5.0μg/kg,在10、20和50μg/kg 3个浓度水平进行添加实验,平均回收率为93.1%~105.5%,批内相对标准偏差为1.5%~4.6%,批间相对标准偏差为1.8 %~5.6%。 关键词 高效液相色谱串联质谱,猪肉,泰拉霉素,残留 泰拉霉素(Tulathromycin)是一种新近上市且为动物专用的大环内酯类半合成抗生素,分子式为C41H79N3O12(分子量806)。我国农业部在2008年第957号公告中首次允许泰拉霉素在动物生产中使用。泰拉霉素主要用于放线杆菌、支原体、巴氏杆菌、副嗜血杆菌引起的猪、牛的呼吸系统疾病。具有用量少、一次给药、低残留和动物专用等众多优点。在我国,大环内酯类药物现行使用较为广泛的是泰乐菌素和替米考星,虽然这2种药物在生产中都取得了良好的效果,但随着使用时间的延长,我国很多地区出现了不同程度的耐药性,导致用量不断增大,但治疗效果却在逐步降低 ,而泰拉霉素药效均强于泰乐菌素和替米考星等市场广泛使用的大环内酯类药物。因此,泰拉霉素在畜禽生产中使用前景非常广阔,其残留分析方法研究也显得尤为必要。 目前国内外大环内酯类药物残留检测方法已有ELISA筛选法、薄层色谱法、气质联用法、高效液相色谱和高效液相色谱串联质谱法等众多方法。但泰拉霉素的残留检测方面研究较少,国内尚未见有相关报道。本研究建立了以罗红霉素(C41H76N2O15, 837)为内标的泰拉霉素HPLC-MS/MS方法,方法的定量限为10μg/kg,可以满足国内外有关法规对其残留检测的要求,可为残留监控提供技术支持。1 材料与方法1.1仪器与材料 Waters 2695 Quattro MicroTM API高效液相色谱串联质谱仪(美国Waters公司);固相萃取仪(美国SUPELCO公司)。 泰拉霉素标准品(Sigma公司);罗红霉素 (Sigma公司);乙腈(色谱纯);甲醇(色谱纯);其余试剂均为分析纯试剂。PCX固相萃取柱(3 mL, 60 mg,AGELA公司)。1.2标准溶液的配制 泰拉霉素和内标储备液:分别准确称取10mg标准品于10mL容量瓶中,用0.05M K2HPO4/乙腈(75:25,v/v; pH6)定容,混匀后置冰箱冷藏储存,有效期3个月。 标准工作溶液的配制:吸取储备液1mL于100mL容量瓶中并用0.05M K2HPO4/乙腈(75:25,v/v; pH6)定容,再从中吸取1mL于50mL容量瓶中得0.2mg/L,用于添加(置冰箱冷藏储存,有效期2个月)。内标物罗红霉素的添加溶液配制方法同泰拉霉素。1.3样品制备 称取2.0g组织样品于50mL的聚四氟乙烯塑料管中,加入10mL甲醇+0.1%磷酸提取液(70:30 v/v)和50μL内标工作液,匀质1 min后5000rpm转速离心2min,收集上清液过已经分别用3mL甲醇和3mL提取液润洗过的60mg/3mL的PCX小柱,待全部过柱后,再用3mL水、3mL甲醇淋洗,最后用4mL4%的氨化甲醇洗脱,50℃水浴氮气吹干后用1mL流动相定容,进行HPLC-MS/MS分析。1.4仪器分析条件 液相色谱条件:Symmetry®C8 5μm 3.9mmx20mm,流动相:乙腈:0.1%甲酸水溶液=70:30;柱温:30℃,进样室温度15℃,进样量:10μL,流速为0.3mL/min。质谱条件:电离模式:ESI(+);检测方式:多级反应检测(MRM);毛细管电压:4.2 KV;锥孔电压:20 V;RF透镜电压:0.2 V;离子源温度:110 ℃;脱溶剂气温度:350 ℃;锥孔气流速:100 L/h;脱溶剂气流速:600 L/h;二级碰撞气:氩气。1.5添加回收率实验 添加回收率实验添加浓度为10、20和50µg/kg,实验步骤同1.3。2 结果与讨论2.1样品的提取和净化 泰拉霉素含3个氨基基团,为弱碱性化合物,易溶于酸性溶液和极性溶剂中,在pH 6~8的水溶液中较稳定,在pH 9条件下均不稳定。对泰拉霉素残留的提取和净化方法的设计主要依据其弱碱性、脂溶性和酸碱不稳定性。本实验比较了4种提取液:乙腈-0.1%偏磷酸(70:30 v/v)、甲醇-0.1%偏磷酸(70:30 v/v)、乙腈-0.1%磷酸(70:30 v/v)、甲醇-0.1%磷酸(70:30 v/v)对泰拉霉素的提取效果。结果表明,甲醇-0.1%磷酸(70:30)提取液对泰拉霉素的提取效果较好,回收率高于其他提取液,故本方法选择甲醇-0.1%磷酸(70:30)为样品提取液。 液-液分配(LLE)和固相萃取(SPE)是大环内酯类药物的2种主要净化手段,其中LLE操作较麻烦且回收率较低、重复性较差,所以本研究采用固相萃取技术净化,并比较了3种SPE净化小柱:SUPELCO C18小柱、Oasis MCX小柱和PCX小柱, 由于Oasis MCX小柱和PCX小柱兼有阳离子交换和疏水作用机制,基于泰拉霉素的弱碱性和脂溶性的理化特性,Oasis MCX小柱和PCX小柱的净化效果好于C18小柱。Oasis MCX小柱和PCX小柱提取效率没有明显差别,但PCX小柱成本低,所以,本方法净化选择PCX小柱。2.2色谱质谱条件的优化[fo
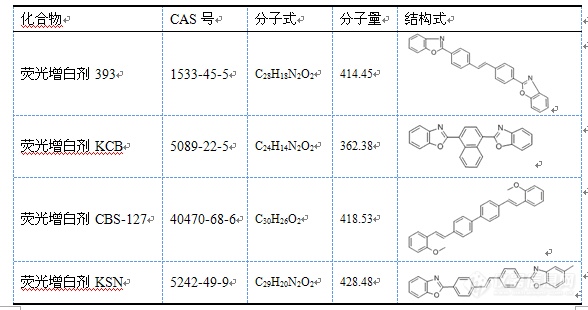
[color=#444444]如题,我想用液相色谱串联质谱检测几种荧光增白剂类物质,用的waters TQ的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url],电喷雾离子源,正离子模式,物质都没什么响应,母离子都看不到,麻烦大家能帮我分析一下吗?物质如下图:[/color][color=#444444][img=,588,310]https://ng1.17img.cn/bbsfiles/images/2019/07/201907251042350788_3973_1752342_3.png!w588x310.jpg[/img][/color]
:高效液相色谱_串联质谱法测定酸奶中纳他霉素的含量期刊:中国卫生检验杂志作者:李媛媛; 王伟; 关树文; 李刚; 董巧红;链接:http://202.119.208.220:8002/kns50/detail.aspx?dbname=CJFD2010&filename=ZWJZ201004027
【序号】:1【作者】:周杨; 冯群科; 朱永林;【题名】:高效液相色谱-串联质谱法测定饲料中三聚氰胺【期刊】:中国饲料【年、卷、期、起止页码】:2010年 12期 【全文链接】:http://202.119.208.220:8002/kns50/detail.aspx?dbname=CJFD2010&filename=SLGZ201012013

液相色谱-串联质谱条件测定水果中的多农药残留方法优化本法建立了水果多种农药残留量的快速、简便、准确的测定方法,通过对样品前处理方法、仪器检测方法的考察优化,建立液相色谱-串联质谱检测方法。其中以日本制定的“肯定列表”中的“一律标准”最为严格,限量为 0.01mg/kg,而我国的残留限量标准还不够完善,很多农药还没有制定限量标准,其方法的测定低限(定量限)能达到 0.01mg/kg的要求。1、液相色谱条件考察:在方法建立过程中对液相色谱条件进行了考察,主要考察了色谱柱、流动相等。在色谱柱选择时,比较了BEH C18、 HSS T3、 Zorbax Eclipse Plus C18等色谱柱,发现HSS T3色谱柱对甲胺磷、乙酰甲胺磷等大极性农药的色谱保留效果较好,所以选择HSS T3色谱柱进行下一步的研究。在流动相考察时,发现在流动相中加入0.1%的甲酸可以改善多菌灵、噻菌灵等农药的分离,而且加入甲酸可以在电喷雾正离子( ESI+)模式电离时提供H+,提高电离效果,所以选择在流动相中加入0.1%的甲酸,比较了在水相和有机相中均加入0.1%的甲酸、仅在水相中加入0.1%的甲酸两种情况,发现色谱分离及质谱电离无显著差别,为简化操作和便于使用,选择仅在水相中加入0.1%的甲酸。流动相的有机相选择时考察了甲醇、甲醇-乙腈( 1+1, V/V)、乙腈三种情况,发现采用乙腈时色谱柱的柱压较低,色谱分离也较好,但毒死蜱、辛硫磷、特丁硫磷、敌敌畏等常用有机磷农药的质谱响应值低、重现性差,而选用甲醇时可以显著提高这些化合物的质谱响应及重现性,综合考虑后选择甲醇为流动相的有机相。由于此次分析的多农药的化学性质差别较大,从高极性到低极性均有分布,所以色谱分离时需要采用梯度洗脱模式,通过实验考察,最终确的液相色谱条件如下:a)色谱柱: HSS T3柱,长100 mm,内径2.1 mm,粒径1.8 μm,或相当者; b) 流动相:甲醇-0.1%甲酸溶液梯度洗脱,参见表 1。 https://ng1.17img.cn/bbsfiles/images/2020/09/202009211516176216_1219_2166779_3.png!w607x264.jpgc) 柱温: 35 ºC
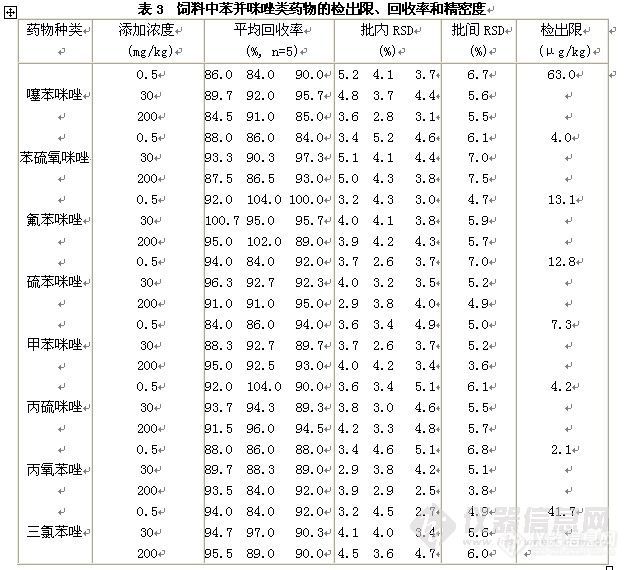
液相色谱串联质谱法测定饲料中8种苯并咪唑类药物摘 要 建立了同时测定饲料中8种苯并咪唑类药物(噻苯咪唑、丙硫咪唑、硫苯咪唑、苯硫氧咪唑、氟苯咪唑、甲苯咪唑、丙氧苯唑和三氯苯唑)的液相色谱串联质谱分析方法。饲料样品直接用酸化乙腈提取,提取液用甲酸溶液稀释后直接进行分析。分析时采用XBridgeTM C18色谱柱,以甲酸溶液-乙腈体系进行梯度洗脱,MRM方式测定,基质外标法定量。苯并咪唑类药物在0.02~10 mg L-1浓度范围内呈良好的线性,线性相关系数均大于0.990,苯并咪唑类药物在饲料样品中最低检测限为2.1~63.0μg/kg。饲料中苯并咪唑类药物在0.50~200 mg/L范围内的回收率为84.0%~104%之间,相对标准偏差(RSD)均小于10%。 关键词 苯并咪唑类药物;液相色谱串联质谱法;饲料 苯并咪唑类药物(benzimidazoles, BMZs)属于广谱、高效、低毒抗蠕虫药,由于对胃肠线虫具有很强的驱杀作用,至今仍在广泛使用。但由于BMZs在实验动物和靶动物显示致畸和致突变作用,目前使用的BMZs多数是食品残留中重要的监控对象,且BMZs在体内转化的代谢产物仍具有毒理作用,所以我国以及联合国粮农组织、欧盟、美国、日本等国家和组织都将苯并咪唑类药物列入限制使用的兽药药物,并制订出各种苯并咪唑类药物在不同动物体内(肌肉、组织、奶等)的最高残留限量。饲料安全直接关系到动物性食品的安全,考虑到苯并咪唑类药物经常被添加到饲料中使用,故很有必要进行饲料中苯并咪唑类药物的分析研究。 目前对于动物组织中苯并咪唑类药物的分析方法较多,而饲料中苯并咪唑类药物分析方法国内未见发表,国外也较少,涉及的种类也较少,最多的仅有5种药物。动物组织和饲料中BMZs分析涉及的主要分析手段有:酶联免疫吸附法( ELISA) 、气相色谱-质谱法(GC-MS)、高效液相色谱法(HPLC)及高效液相色谱串联质谱法(HPLC-MS/MS),高效毛细管电泳法(HPCE)。考虑到苯并咪唑类药物在我国使用情况,本研究选择了8种常用苯并咪唑类药物,考虑到LC-MS/MS法灵敏度高的特点,样品酸化乙腈提取后直接稀释后进行液相色谱串联质谱分析。1 材料与方法1.1 仪器与试剂 Waters 2695 Quattro MicroTM API高效液相色谱串联质谱仪(美国Waters公司),配置电喷雾离子源;固相萃取仪(美国Supelco 公司);Sigma离心机。噻苯咪唑和丙硫咪唑标准品(Accustandard 公司);硫苯咪唑、苯硫氧咪唑、氟苯咪唑、甲苯咪唑、丙氧苯唑和三氯苯唑标准品(Dr. Ehrenstorfer)。乙腈、二甲亚砜和甲酸为色谱纯(Fisher公司)。1.2 仪器条件 XBridgeTM C18色谱柱(150 mm×2.1 mm,内径3.5 μm);流动相A为0.1%甲酸溶液,B相为乙腈,梯度洗脱条件:B相在1.0 min内从15%线性增加到25%,再在2.5 min内线性增加到95%,保持3.5 min,然后在0.1 min内降至15%,保持4.9 min;流速:0.3 mL/min;进样量:10 µL;柱温:30℃。 质谱条件:ESI源正离子模式电离;多反应监测(MRM);毛细管电压:3.0 KV;萃取锥孔电压:20 V;RF透镜电压:0.5 V;离子源温度:110 ℃;脱溶剂气温度:350 ℃;锥孔气流速:50 L/h;脱溶剂气流速:600 L/h;倍增器电压:650 V;二级碰撞气:氩气;其它条件详见表1。http://ng1.17img.cn/bbsfiles/images/2010/11/201011301506_262957_1759541_3.jpg1.3 样品处理 称取2g试样(精确到0.01g)于50 mL离心管中,加入20 mL0.5 %甲酸乙腈,涡旋1 min,然后超声提取10 min,以5000 r/min的速度离心5 min后吸取1.0 mL上清液于5 mL刻度试管中,加入3 mL0.1 %甲酸溶液于试管中,混匀后过0.22 μm滤膜,进行液相色谱串联质谱分析。1.4 线性实验 准确称取各10.0 mg BMZs标准品于相应的10mL容量瓶中,噻苯咪唑、甲苯咪唑、丙氧苯唑和丙硫咪唑用二甲亚砜溶解并定容至刻度,其余4种BMZs用甲醇:二甲亚砜(2:3 v/v)溶解并定容至刻度,即得均为1000 mg/L标准储备液。分别吸取1.0 mL各标准储备液于同一10mL容量瓶中,用甲醇稀释至刻度,即得100 mg/L的混合标准工作液。分别准确移取苯并咪唑类药物混合标准中间液适量,配制浓度为0.2.、0.8、2.0、10.0、40.0和100.0 mg/L的系列标准溶液,吸取0.1 mL于5 mL刻度试管中,再吸取空白试料提取液0.9 mL于该5 mL刻度试管中,加入3 mL0.1%甲酸溶液后混匀过膜,进行上机测定,以定量离子对峰面积为纵坐标,标准溶液浓度为横坐标,绘制基质校准标准曲线。2 结果与分析2.1 液相色谱质谱分析 苯并咪唑类药物色谱分析时,通常采用反相分离体系,主要有三类流动相体系:离子增强体系,pH2~3,一般使用乙腈-磷酸或磷酸盐体系;离子抑制流动相体系,pH5~7;离子对流动相体系,离子增强流动相中加入阴离子对试剂。对于多组分苯并咪唑类药物液相色谱质谱分析时,通常采用离子增强体系进行梯度洗脱,如0.1%甲酸溶液-乙腈体系,因为该体系和纯水-乙腈体系相比色谱峰的拖尾现象得到了明显改善。 苯并咪唑类药物属弱碱性物质,中等极性,在酸性条件下很容易质子化,于是本方法选择ESI+进行分析。以乙腈/0.1%甲酸溶液(3:7,v/v)为溶解液,用蠕动泵(20μL/min)对苯并咪唑类药物的质谱条件进行优化。经过优化的条件为:毛细管电压:3.0KV;离子源温度:110℃;脱溶剂气温度:350℃;锥孔气流速:50L/h;脱溶剂气流速:600L/h。其它条件详见表1。2.2 提取净化方法的选择和优化 [font=宋体
请教[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]-质谱/质谱,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]-质谱联用,液相色谱-串联质谱,液相色谱质谱/质谱联用,这几个名称是一样的吗?
超高效液相色谱-串联质谱检测动物源性食品中β-受体激动剂残留分析方法的优化
液相色谱-串联质谱法检测食品中维生素D含量 如何用液相色谱-串联质谱法(LC-MS/MS)检测食品中的维生素D含量。这项技术可是现代食品分析中的佼佼者,能够帮助我们精准地掌握食品中的营养信息。 样品前处理 提取:首先,我们需要从食品样品中提取维生素D。这一步很关键,因为提取效率直接影响最终的检测结果。通常,我们会使用有机溶剂,比如乙腈或甲醇,来提取维生素D。 净化:提取后的样品往往含有很多杂质,这些杂质会影响检测结果。因此,我们需要对提取液进行净化处理。常用的净化方法有固相萃取(SPE)和液液萃取(LLE)。 浓缩:净化后的样品溶液需要进行浓缩,以提高维生素D的浓度。常用的浓缩方法有氮气吹干和旋转蒸发。 仪器操作 流动相选择:选择合适的流动相对分离效果至关重要。通常,我们会使用水和有机溶剂(如甲醇或乙腈)的混合物作为流动相,并根据需要添加少量酸或缓冲液。 色谱柱选择:选择适合的色谱柱也很关键。C18反相色谱柱是常用的选择,因为它对维生素D有很好的保留效果。 质谱条件:设置合适的质谱条件,包括离子源温度、喷雾电压、碰撞能量等。这些参数的优化可以大大提高检测灵敏度和特异性。 故障排除 峰形不好:如果发现峰形不好,可能是由于流动相比例不合适或色谱柱污染。尝试调整流动相比例或清洗色谱柱。 灵敏度低:如果灵敏度不够,可能是由于样品提取效率低或仪器参数设置不当。检查提取方法并优化仪器参数。 杂峰干扰:如果出现杂峰干扰,可能是由于样品净化不彻底或流动相选择不当。尝试改进净化方法或更换流动相。 仪器故障:遇到仪器故障时,首先要保持冷静,然后根据仪器的报错信息查找原因。必要时,可以联系仪器厂家进行维修。 总之,液相色谱-串联质谱法检测食品中维生素D含量是一项复杂但非常重要的技术。通过掌握这些操作要点和故障排除方法,我们可以更加准确、高效地完成检测任务。
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=152834]液相色谱-串联质谱法检测牛乳中多肽类抗生素残留量[/url]摘 要选用牛奶为研究对象.建立了一种可同时测定多粘菌素B、粘杆菌素、杆菌肽和维吉尼霉素等4种多肽类抗生素残留量的液相色谱一串联质谱法(Lc—Ms/Ms)检测方法。样品经甲醇一0.1%甲酸体系提取,正己烷脱脂,固相萃取柱净化后,利用Lc—Ms/Ms进行定性和定量分析。结果表明,该方法4种多肽类抗生素检出限分剐为:多粘茵素B 25O g/kg,粘杆菌素25 g/kg,杆菌肽A 5O g/kg,维吉尼霉素2O g/kg。平均回收率分别为:多粘茵素B 92.16%—95.89%,粘杆菌素92.24% 97.87%,杆菌肽94.54%—97.96%,维吉尼霉素 93.58% 8.25%。变异系数为2-54^ .55。
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=152868]深海鱼油中脂肪酸的柱前衍生-高效液相色谱串联质谱分析[/url]摘要:用1.【2.(对甲苯磺酸酯)乙基卜2.苯基咪唑【4,5一f】9,10一菲(TSEPIP)作为柱前荧光衍生试剂,在Eclipse XDB.Cs(4.6× 150mm,5lam,Agilent)反相色谱柱上,采用梯度洗脱在检测波长为380nm(激发波长为260nm)的条件下,实现了阿拉斯加深海鱼油中饱和脂肪酸含量的外标法定量测定。26种饱和脂肪酸的线性范围是200.0pmol48.83fmol,线性相关系数均大于0.9996,检测限为3.824~47.13fmol(信噪比为3:1测得,S/N 3:1)。经柱后串联质谱大气压化学电离源(APCI)正离子模式实现了各种饱和与不饱和脂肪酸衍生物的质谱鉴定,进而通过峰面积归一化法得出了所有饱和与不饱和脂肪酸的相对含量。结果表明,深海鱼油主要含有C12~C22的脂肪酸,共鉴定出25种脂肪酸,其中不饱和脂肪酸含量占69.71%(峰面积百分比,下同),特别是具有重要生理作用的多不饱和脂肪酸,如C2o:5:5,8,11,14,17一二十碳五烯酸(5,8,11,14,17一eicosapentaenoic acid,EPA,16.62%),C22:6"2,5,8,11,14,17一二十二碳六烯酸(2,5,8,11,14,17.docosahexenoic acid,DHA,12.31%)。
GB/T 23584—2009《水果、蔬菜中啶虫脒残留量的测定 液相色谱-串联质谱法》[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=180639]GBT 23584-2009 水果、蔬菜中啶虫脒残留量的测定 液相色谱-串联质谱法.pdf[/url]
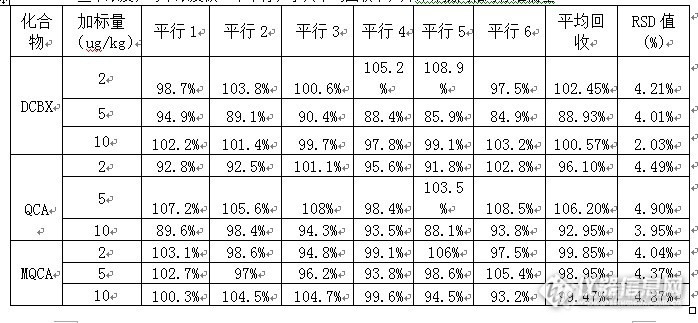
液相色谱串联质谱法测定动物组织中卡巴氧和喹乙醇代谢物残留量 摘要:采用高效液相色谱-电喷雾串联质谱仪(LC-ESI-MS-MS),建立了猪肉中卡巴氧代谢物脱氧卡巴氧、喹恶啉-2-羧酸和喹乙醇代谢物3-甲基喹恶啉-2-羧酸残留量的药物残留的检测方法,样品用甲酸溶液消化,蛋白酶水解,盐酸酸化,离心过滤后,过Oasis MAX固相萃取住或相当者净化。先用二氯甲烷洗脱脱氧卡巴氧,再用%甲酸乙酸乙酯溶液洗脱喹恶啉-2-羧酸和3-甲基喹恶啉-2-羧酸,氮气吹干洗脱液,残渣用甲酸+甲醇(19+1)溶液溶解,样液供液相色谱-串联质谱仪测定,内标法定量。本方法采用了2ug/kg,5ug/kg,10ug/kg,3个添加浓度,每个浓度6个平行样品,上述3种药物残留的回收率在80%~110%,相对偏差在2.03%~4.94%。关键词:液相色谱串联质谱法;脱氧卡巴氧;喹恶啉-2-羧酸;3-甲基喹恶啉-2-羧酸。1 引言卡巴氧(Carbadox) 和喹乙醇(Olaquindox) 同属喹喔啉类化合物 , 该类药物具有显著的促进动物生长的作用 , 用作猪等养殖动物的饲料添加剂。二者本身具有潜在的致畸变、致癌作用 , 其代谢物也可能带来健康风险。因此许多国家将以卡巴氧和喹乙醇列为对食用动物禁用或限用的药物 , 欧盟、中国、日本、美国、澳大利亚等对二者在动物组织内迅速代谢而产生的相应的代谢产物喹喔啉-2-羧酸(QCA)和 3-甲基喹喔啉-2-羧酸(MQ-CA)制定了残留监控的限量标准。在动物体内,喹乙醇和卡巴氧、经脱单氧、脱双氧后主要生成脱氧卡巴氧、喹恶啉-2-羧酸、3-甲基喹恶啉-2-羧酸,相对应的代谢物比较稳定,通常作为残留分析和监控的目标物质,代谢途径见图1。鉴于喹乙醇和卡巴氧的毒性和潜在的危害,为了更好的对动物食品进行监控,本文旨在建立喹乙醇、卡巴氧代谢物的残留液质联用仪检测方法。 http://ng1.17img.cn/bbsfiles/images/2013/10/201310242215_472741_2082444_3.jpg2 实验部分1.1仪器与试剂1.1.1试剂和材料甲醇:德国默克,色谱纯。乙腈:德国默克,色谱纯;乙酸乙酯:德国默克,色谱纯;水:1.25L哇哈哈纯净水(杭州产);正己烷:Honeywell,色谱纯。甲酸:色谱纯乙酸:色谱纯浓盐酸:分析纯乙酸钠:分析纯甲酸乙酸乙酯溶液:2% 向400mL乙酸乙酯中加入10mL甲酸,用乙酸乙酯定容至500mL。甲酸溶液







 我要推广仪器
我要推广仪器
 下载APP
下载APP