【专家讲座】:液相色谱三重四极杆质谱联用技术在水中低浓度有机物分析领域的应用【讲座时间】:2015年05月14日 14:00【主讲人】:向华:上海市供水调度监测中心水质监测站(国家城市供水水质监测网上海监测站)高级工程师,长期从事水质分析工作,主要负责水中有机物的检测、新增检测方法的开发,同时负责实验室的质量控制工作。【会议简介】本报告通过介绍水中有机污染物的种类、目前常用的检测方法和在检测过程中存在的问题,引入了液相色谱串联二级质谱方法在水中低浓度有机污染物分析领域的应用。阐述了该检测方法具有灵敏度高、定性准确、水样前处理简单、分析周期短等优势,是水质分析中应该优先选择的检测方法。-------------------------------------------------------------------------------1、报名条件:只要您是仪器网注册用户均可报名参加。2、报名截止时间:2015年05月14日 13:303、报名参会:http://www.instrument.com.cn/webinar/meeting/meetingInsidePage/14364、报名及参会咨询:QQ群—379196738
液相色谱有没进样最低浓度?我昨天做的估计浓度太低了,大约0.4ug.ml,没有任何峰出现,就一条直线
液相色谱经常在空白和低浓度对照溶液中出现异常峰,不同仪器不同色谱柱,不同产品,都是经常出现不规则鼓包峰,求助各位大神
[color=#444444]液相色谱高浓度时为单峰,低浓度时出现两个峰,且两个峰没有达到基线分离,怎么办? 试了中间浓度,峰形为一个大峰的右翼上出现一个小峰。怎么办呢,新手求助。[/color][color=#444444]等度洗脱水(0.05%甲酸):乙腈=35:65,样品用乙腈溶的,样品易溶于有机相,水中溶解性差[/color]
液相色谱做多菌灵标准品,低浓度杂峰少,高浓度杂峰多,是什么原因[img]http://ng1.17img.cn/bbsfiles/images/2017/11/201711131029_01_3163820_3.jpeg[/img][img]http://ng1.17img.cn/bbsfiles/images/2017/11/201711131029_02_3163820_3.jpeg[/img][img]http://ng1.17img.cn/bbsfiles/images/2017/11/201711131029_03_3163820_3.jpeg[/img]
做一批样品时,浓度肯定有的高有的低,我们都是不知道哪个高哪个低的,如果把高的排在前面,低浓度的排在后面的话,会不会对低浓度的样品有影响,好像其它机器都有一瓶空白液,可以设定每做完一个样品,清洗一次管路的,岛津LC-20A液相好像没有这个功能?
液相色谱分析中标准样的浓度一般是多少?我是混合标准样
请教一下,我用的是东西电子的液相色谱仪,用万分之一浓度萘甲醇标液进样,不出峰是什么原因?
用高效液相色谱仪测定农药残留的呋喃丹,色谱柱是C18的15厘米短柱,使用的是荧光检测器,波长339和445,柱后衍生流速都是0.5,水解温度95度,柱温42度,流量1。流动相是水和甲醇的梯度,使用此方法能检测到100ng/ml的标样,检测不到40ng/ml的,我通过改变流速,增大进样量,以及增加柱温都没有什么效果,另外荧光检测器的基线峰波动在700左右,柱后衍生的工程师认为低浓度不出现是因为被基线峰淹没了,求大神指点怎样降低极限峰值或者能使样品峰出的更明显,另外检测草甘膦也遇到了同样的问题,我们的检测器是赛默飞U3000,荧光检测器单位是counts,不是电压单位
(1)液相色谱柱色谱柱使用前注意事项:液相色谱柱的储存液无特殊说明,均为评价报告所示的流动相。在使用前,一定要注意液相色谱柱的储存液与要分析样品的流动相是否互溶。在反相色谱中,如用高浓度的盐或缓冲液作洗脱剂,应先用10%左右的低浓度的有机相洗脱剂过渡一下,否则缓冲液中的盐在高浓度的有机相中很容易析出,堵塞色谱柱。(2)流动相:流动相中所使用的各种有机溶剂要尽可能使用色谱纯,配流动相的水最好是超纯水或全玻璃器皿的双蒸水。如果将所配得流动相再经过0.45μm 的滤膜过滤一次则更好,尤其是含盐的流动相。另外,装流动相的容器和色谱系统中的在线过滤器等装置应该定期清洗或更换。以常规硅胶为基质的键合相填料通常的pH值适用范围是2.0-8.0。当必须要在pH值适用范围的边界条件下使用色谱柱时,每次使用结束后立即用适合于色谱柱储存并与所使用的流动相互溶的溶剂清洗,并完全置换掉原来所使用的流动相。(3)样品:样品也要尽可能清洁,可选用样品过滤器或样品预处理柱(spe)对样品进行预处理;若样品不便处理,要使用保护柱。在用正相色谱法分析样品时,所有的溶剂和样品应严格脱水。
液相色谱的市场分析就目前的市场情况来看,国产[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的需求要远远大于国产液相色谱。这并不是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的市场需求量大于液相色谱,而恰恰相反,在未来几年[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的需求会逐渐变小,而液相会呈上升趋势。这主要原因是国产的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]已被国人认可,认为常规分析国产[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]完全能胜任。而液相色谱还是违背国内分析人士认可,认为稳定性差、故障率高等缺点。所以我们还需走较长的路,但我们看好这市场,相信好的产品最终是会被市场认可的。 首先是如何看指标。液相色谱仪的指标很多,有泵的、检测器的、色谱柱等等。我们认为要看主要技术指标,根据国家标准,仪器的主要指标有噪音,漂移,最小检测浓度,定性定量重复性等。这些指标都要放在系统,回路里去看,去比较。就是需要把各单元装置都要联接好,如接好色谱柱,进样阀,并且要通上流动相。因为您在分析中也都是联接好以后才可以进行分析的,而不是单单用个检测器或泵的。然后在这个基础上,我们再去比较这些主要指标。1.噪音是指由仪器的电器元件、温度波动、电压的线性脉冲以及其他非溶质作用产生的高频噪声和基线的无规则波动。噪音的大小直接关系到仪器的检测灵敏度,噪音越大,检测的灵敏度就越低。对于检测低含量的样品就要求仪器的噪音越小越好,否则噪音过大将会导致基线不稳,进而影响分析结果。2.最小检测浓度(最小检测限):是反映仪器灵敏度的重要参数。CL=2×Nd×C/H(CL :最小检测浓度 Nd:噪音 C:样品浓度最小检测浓度 H:样品峰高)由上式可见,最小检测浓度是和噪音成正比的,噪音越大,最小检测浓度就越大,灵敏度就越低。某些厂家回避了这个指标,说明他们不愿在最小检测浓度的基础上去比较噪音。这里来解释一下:最小检测浓度是考验仪器的灵敏度。最小检测浓度数值大,仪器的灵敏度就小,不能反应真正的噪音和漂移水平。例如:我公司的最小检测浓度小于1×10-8g/ml (萘/甲醇溶液),而某些仪器是:4×10-8g/ml (萘/甲醇溶液)。这就说明我们的仪器灵敏度大,可以检测更微量的样品。同样如我公司仪器把最小检测浓度调较得和其它产品是一样,也就表明我们可以来得比其它产品噪音和漂移低4倍。可以从这个公式就可看出:最小检测浓度=2*仪器的噪音*进样的样品浓度/样品的峰高值那光程又代表什么?我们先看下面一个公式,比尔定律: A=log(I0/I)=εCLA是吸收率;I0代表参照池的光强;I为样品池的光强;ε为摩尔吸光系数;C是样品浓度;L就是流通池的光程。 可以看出在同样的“C”样品浓度情况下,“L”流通池的光程越大,仪器的“A吸收率”也就越大。这样可以检测到的样品浓度就越小。为什么有些厂家把仪器的流通池光程做得很小,有些只有:3.5mm、4.5mm或5.5mm,而不是我公司的8mm。这是因为这些厂家不能有效的降低整个系统的“噪音和漂移水平”,只能牺牲流通池光程,也就是牺牲了仪器的检测灵敏度和最小检测浓度,来达到降低“噪音和漂移水平”目的。用通俗的说法比喻:HPLC就是台音响,光程就是这台音响的音量控制键,光程越小就是把音量调小了,耳朵对音响本身的噪音和失真(可以理解为仪器的噪音和漂移)感觉就越小。但也就说明了这些仪器整体系统的制造水平不高。3.漂移是指仪器稳定后一段时间内基线漂离原点的距离,通常用来衡量仪器稳定快慢。高品质的仪器能在较短的时间内达到稳定,从而在一定程度上提高了分析效率。 4.定性定量重复性主要是考核仪器稳定性的指标,这对于分析样品来说是非常重要的。好的仪器其稳定性应该是十分优秀的,这就要求多次进样保留时间及含量的一致性,这样做出来的结果才能使人信服。有的朋友会认为这些指标好像是都是检测器的。对的,但是就前面所说的是要放在整个回路和系统里去看去比较。例如:泵的脉动会直接影响噪音指标,泵的流量准确度、精确度指标,以及密封性不好也会影响相关指标。所以要系统地看指标。例如:某些公司在公布的指标中,噪音和漂移指标写的条件是空池或有的干脆不写。这个指标只考核了UV检测器光学和电气的特性,与实际情况相差甚远,并没有考验泵的压力脉动,液流回路的阻尼和UV检测器流通池的性能。所以我们认为从以上的主要指标中可以反应出仪器的一些真实水平。二.操作方便:操作方便性无论是对新手还是成熟的用户都是很重要的。操作越简单,有利于提高分析效率,也为以后分析方法的拓展提供有力的帮助。我公司的WS-100工作站软件实现了真正的智能化操作。它摒弃了以往工作站软件只能起到数据处理的理念,率先在国内推出了集控制与后处理于一身的软件,在操作使用上给用户带来了极大的方便。三.系统性这里指的是仪器的整体开发,是一个完整的系统。目前市场上有这个现象,说我的泵是进口的,或者是检测器怎么好,用了什么很多的进口件组装等等。其实这是个误区。液相色谱是个复杂的系统,不是整体开发的,各项指标之间、软件和硬件、硬件和硬件等都不匹配,整体水平不会高到哪里去。而且在售后服务方面对用户也是不负责任的。四.稳定可靠,故障率低五.适合的才是最好的选择适合自己的产品,并不是价格越贵就越好。在满足实际使用的前提下,性价比往往应是需要考虑的因素。
液相色谱分析是指流动相为液体的色谱技术,是色谱法中最古老的一种,但通过改进填料的粒度及柱压,在经典的液相柱色谱的基础上引入了[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的塔板理论,在技术上采用了高压输液泵,高效固定相和高灵敏度的检测器,实现了分析速度快、分离效率高和操作自动化,这种色谱技术被称为高效液相色谱法(HighperformanceliquidchromatographyHPLC)。中药的成分非常复杂,以往常用的薄层色谱等方法因其精密度、准确度、灵敏度、重现性差而不能满足现代中药的需要。高效液相色谱正是以其稳定、可靠、高效的特点成为中药研究的最重要的分析方法。目前高效液相色谱已经广泛应用于生物碱、皂苷、黄酮、蒽醌、香豆素等各种中药有效成分的测定。近年来对高效液相色谱监测中药的研究非常多,由于高效液相色谱集经典液相色谱和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的优势于一身,无论柱效、选择性还是分析程度都达到或超过了它们,近年来对高效液相色谱的不足之处进行了改进,使这项技术日臻完善。 1高效液相色谱发展近况 高效液相色谱在药物分析中的应用,主要考虑试样的预处理和分析柱、检测器的选择。在试样的预处理上,日前兴起的固相(微)萃取使得许多含量低的成分得到精制提纯,从而适于高效液相色谱的测定,而孙新国采用逆流萃取测定川芎嗪含量取得了很好的效果。中药中有些紫外吸收弱,或无特征紫外吸收的成分,直接用高效液相色谱测定,其灵敏度和分离度都不尽人意,利用柱前或柱后衍生化法可使这些成分较精确地测定出来。对于极性大、脂溶性差物质,在YWGCl8柱上不易保留,用十二烷基磺酸钠作为离子对试剂,降低其极性,延长柱上的保留时间,取得较好的分离较果。将液相色谱和质谱这两个强有力的分析技术在线连接在一起,经过三十年的发展已成为一项较为成熟的分析手段,但是它从形成伊始就存在着问题:从液相色谱流进质谱时,流动相的变化、溶剂的组成、高温高压离子化的问题制约着这种联用技术发展,大气压离子化接口具有去除溶剂和离子化的双重功效,它的引入,使得该技术在各个领域得到了广泛的应用。电喷雾离子源是一种软电离技术,一般只生成(M+H)+和(M-H)-两种分子离子峰,选择性监测(mz)190的负分子离子峰,具有较高的灵敏度、准确度、专一性,满足了低浓度药物研究的需求。由张莉等人研究的三维高效液相色谱法可以同步测定葛根素和阿魏酸两种指标。通过实验证明:如果选择合适的柱温等色谱条件,乙醇作为反相高效液相色谱流动相,分析中药及中成药中有效成分,既安全又准确。结构相似的物质,普通的检测器难以检测出来,高效液相色谱-电化学法可以有效地测定黄连粉中仅差一个基团的黄芩苷和黄芩素的含量。样品经色谱柱分离后收集,再经荧光分光光度计测荧光强度,影响因素多,测定复杂,改进后的高效液相色谱-荧光法则可以不经衍生化和收集分离物,只经化学处理除杂,浓缩后直接进样即可。用该法测定贯叶连翘中金丝桃素的含量也取得了较好的结果。高效液相色谱-示差折光测黄芪精口服液中黄芪甲苷的含量也都取得了较为满意的结果。对于只有紫外末端吸收,用紫外检测时灵敏度低,基线易漂移,示差折光检测其易受外界条件干扰,蒸发散射检测器能克服以上不足,响应值只与样品质量有关,其信号相应与质量成正比,不同化合物,质量相同则信号相应基本一致。蒸发光散射检测法是基于不挥发样品分子对光的散射程度与其质量成正比,与其所含基团性质无关。只要选择适当的检测器参数,便可使流动相和溶剂完全气化,不产生信号,而样品中的各个组分以不挥发粒子存在,对光有散射,以被检测出来。因此,蒸发光散射检测器可用于含不同基团的多组分同时分离、分析。和紫外、荧光等方法相比,蒸发光散射检测法对不同物质有近似相同的响应因子, 因而不出现低浓度、高响应或高浓度、低响应的现象,有利于不同比例混合物的准确测定.高效液相色谱-蒸发光散射检验法测定银杏叶中萜类内酯含量、红参及育精胶囊中人参 皂苷Rg1和Re的含量和藤黄中藤黄酸含量都得到了很好的结果。 2高效液相色谱的研究动向 2.1缩短分析时间,提高分离效率。应用先进的检测仪器和方法,对流动相、固定相进行调节或改变,采用梯度洗脱、柱切换技术有望解决这个问题。梯度洗脱的高效液相色谱法,能分析较宽极性范围的样品,较等度洗脱具有很大的优势,但对于成分更复杂、极性范围更宽的中药样品则有些力不从心。多柱高效液相色谱法又称多维高效液相色谱法。除具有梯度洗脱一样的改变流动相浓度的优点外,还可以改变固定相种类、键合度、粒径、柱长、柱径等及流动相种类、浓度等。 2.2进一步向自动化、智能化及联用技术上发展。液相色谱与质谱联用在国外已成为测定低浓度生物药品中药物及代谢物的首选方法,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]-MS法测定血浆中HIV-1蛋白酶,准确高效,血浆中残留的内源性组份和其他药物不干扰测定,既节省材料又节约时间。已经应用于体液、血浆、血清中的药物分析。中药复方注射液“清开灵”中的胆酸类物的分析采用液相色谱质谱质谱联用,效果理想。高效液相色谱-核磁共振联用在药物分析方面的作用很不错。新近提出的智能多柱高效液相色谱系统利用切换技术的模块式分离性能,把样品分块的切换进不同性质的色谱柱,再用合适的流动相洗脱。全过程采用智能化控制。 3高效液相色谱在中药分析中的应用前景 中药研究的大趋势是全成分分析,通过对从单味药到复方的不同配伍、煎煮时间等的研究,才能发现中药中化学成分的变化规律,找到中药机理之间的有机联系。中药成分繁多,且各种成分的性质遍布所有极性段、酸碱范围。实现多成分分析的最简单途径即在一根足够长的色谱柱上,采用温和的流动相,在足够久的时间内洗脱。但这与现代分析要求的简便快速相违。通过大量的应用研究表明,高效毛细管电泳在分析中药成分,尤其在分析高极性化学成分方面有较大优势,在分析大量的复方制剂方面显示了较高的能力。由于毛细管几乎不会出现高效液相色谱分析中常出现的柱床污染现象,而且用过的毛细管柱只需很短的时间进行冲洗后,即可以进行第二个样品的分析,快速高效且分辨率很高。新兴的毛细管电色谱是集高效液相色谱和毛细管电泳优势于一身的一种新型电分离微柱液相色谱技术,它是将高效液相色谱的多种填料微粒移到毛细管中,以样品与固定相间的相互作用作为分离机制,以电渗流为流动相驱动力的色谱过程。最近,一些先进的检测仪器成功的用在了高效液相色谱分析法上,使得高效液相色谱的应用更广泛,并充分利用高效快速、选择性好、灵敏度高等优点,建立更加系统的成分分析方法。通过与质谱联用、梯度洗脱、柱切换技术、配合先进的检测技术,以及与分子生物学、现代分子药理学相结合,必将在中药分析中发挥很大作用。
一、液相色谱发展史色谱分析法是分析化学中获得光泛应用的一个重要分支。色谱法最早是由俄国植物学家茨维特(M.S.Tswett)在1906年研究用碳酸钙分离植物色素时发现的,色谱法(Chromatography)因之得名(经典液相色谱法)。后来在此基础上发展出纸色谱法、薄层色谱法、[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法、液相色谱法。30~40年代发展了柱分配色谱和纸色谱。50年代发展了[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法和薄层色谱法。60年代发展了凝胶色谱法及高效液相色谱法70年代发展了高效毛细管[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法。80年代发展了毛细管电泳和电色谱。90年代出现了光色谱。60年代[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]对高沸点有机物分析局限性发展了液相色谱弥补气谱的不足。高效液相色谱High Performance Liquid Chromatography高压液相色谱High Pressure Liquid Chromatography高速液相色谱High Speed Liquid Chromatography高分离度液相色谱High Resolution Liquid Chromatography现代液相色谱Modern Liquid Chromatography二、液相原理:比尔定律:一束平行单色光经过具有紫外线吸收作用的溶液时,透过溶液的光强度不仅与溶液的浓度有关,而且还与溶液的厚度及溶液本身对光的吸收性能有关: A= KCb其中A为消光值,是透射光强I和发射光强I0的比值的对数(反射光强度忽略不计),即A= lg(I0/I);K为某溶液的消光(吸收)系数,一种有色溶液对于一定波长(单色光)的入射光的K值具有一定数值。若溶液的浓度以mol/l表示,溶液厚度以cm表示,则此时的K值称为摩尔消光系数,它是有色化合物的重要特征之一;C为溶液的浓度;b为光程,即溶液的厚度。该定律只适用于单色光和低浓度的有色溶液。三、液相色谱组成:(一)、主件泵、进样阀、色谱柱、检测器、工作站(记录仪)(二)、附件过滤装置、脱气装置、柱温箱、收集装置等等。(三)、工作程序:液体进入泵-压力传感器-脉动缓冲器-进样阀-色谱柱检测器(四)、泵体组成部分:电机、马达、双柱塞串联泵腔、缓冲器、压力传感器、面贴(五)、检测器组成部分:1、电器部分(变压器、氘灯板、系统电源伴、控制板、显示板、前置板、面贴)2、光学部分(氘灯、灯箱、光学盒、凹面镜、分光镜、小参比、单色器、流通池、前置板)

[color=#3e3e3e]在液相色谱分析过程中,我们经常遇到的问题主要有二种,一种与液相色谱仪器本身因素有关,如,液相色谱的阀门、混合器、检测器的光源以及其它的一些硬件设备。出现这类问题后,如果能找出问题根源,解决起来一般很简单,而且这类问题可以通过对仪器的精心维护来避免;而另一类问题则是分析方法本身造成的问题,如,出现色谱峰形状不好、峰与峰之间不能分开、基线飘移等等。不幸的是,如果出现这类问题,看起来似乎很明显,但是要找出原因并解决这类问题却非常困难。为了减少出现这一类型的问题,就必须在分析之前,仔细研究并选择一个好的分析方法,有了一个好的分析方法,就很容易获得理想的分离效果,而且在出现问题是也很便于找出原因。要选择一个好的分析方法,就必须对液相色谱分析的一些基本原则要有一个很深的了解。[/color][color=#3e3e3e][b](1)分析方法选择的基本原则[/b]假如你想做一顿丰盛的晚餐,首先必须看一下食谱,然后检查一下你所需的东西是否齐全,如果少了配料还必须去商店购买,这样你才可能做出一顿可口的晚餐。同样进行液相色谱分析时,也必须按照一定的程序进行,首先你必须要有专门的仪器和试剂,然后有目的地选择分析方法,这样你才可能得到好的分析结果,避免走一些弯路。[/color][color=#3e3e3e][b](2)柱子的选择[/b]现在大部分液相色谱分析都是使用反相色谱柱,其中以C[sub]18[/sub]和C[sub]8[/sub]柱最为流行,然而,色谱分析并不是流行歌曲,这两种色谱柱之所以运用广泛是因为在大多数情况下,使用这两种柱子都能获得理想的分离效果。尽管一些样品的分离并不是这两种柱子(如,表一所示),但C[sub]18[/sub]和C[sub]8[/sub]经常是最好的固定相,C[sub]18[/sub]和C[sub]8[/sub]柱两者之间并无明显的差别。色谱分析人员遇到的多数样品需要利用反相色谱柱进行分析,但一些样品可能需要其它的分析技术才能成功地分离,这些样品及分离方法在表一中作了简要的叙述。[/color][color=#3e3e3e][img=,527,121]http://ng1.17img.cn/bbsfiles/images/2017/05/201705142201_01_3214098_3.png[/img][/color][color=#3e3e3e]表一:样品及分析方法[/color][color=#3e3e3e]在过去的15年中,高纯度、低金属杂质含量的硅胶基质色谱柱是最为实用的色谱柱之一。但传统色谱分析用的硅胶颗粒具有差异性及酸性表面,固定在柱子表面后,导致出现拖尾峰,而且柱与柱之间的重现性较差。最近硅胶基质中出现一种新的产品,这种硅胶具有Metal-free特性,因此,柱子性能更稳定,重现性也更好,分离后峰的形状也很不错。这种改性后的硅胶称为B型硅胶。由于具有上述优点,大多数色谱制造商都用不同的名称来表明它们与传统的产品之间的不同,如,Intertsil (日本)、 BDS(USA)、YMC--Base(USA)等等。大多数色谱柱制造商都生产以B型硅胶为基质的色谱柱,因此你可以选择你所喜欢的供应商提供的产品。由于这种产品具有上述特性,建议使用A型硅胶柱的改为使用B硅胶柱。[/color][b](3)柱子尺寸规格的选择[/b][color=#3e3e3e]液相色谱分析时柱子选择的另一个因素就是柱子大小、及填充颗粒的直径(dp)的选择。最常用颗粒直径为5μm,但颗粒的直径(dp)为3.0及3.5μm的也适合分析使用,但大多数色谱工作者都喜欢使用颗粒直径为5μm的色谱柱,因为这种色谱柱具有很长的使用历吏。颗粒的dp小意味着可以获得较高的理论塔板数,但所需的柱压增大,而且dp为3.0μm的色谱柱易堵塞,所以一些色谱制造商又生产出dp为3.5μm的色谱柱。在实验室中经常使用的是150mm×4.6mm,dp为5μm的色谱柱。另一种可以选择 的色谱柱为75mm×4.6mm,dp为3.5μm的柱子,这种柱子的分离效果与前一种色谱柱分离效果相似,但使用时间减小一半。这两种柱子还有一个优点就是在合理的压力下(2000psi ),流速可以设定在1.5-2.0mL/min之间,而流速高意味着可以缩短分析时间。有些分析者建议使用30 or 50mm长的柱子作为前处理柱,但这种柱子不能提供足够的塔板数。另一种意见是使用窄孔柱(2mmid)或微孔柱(≤1mmid),这两种柱子所需的溶剂少,但是这两种柱子所需的某些仪器与正常使用的色谱仪有些不同,而且正处在一个研究阶段,建议等这两种色谱柱研究成熟之后再使用。[b]150mm×4.6mm,dp为5μm的色谱柱最为常用,75mm×4.6mm,dp为3.5μm的色谱柱也中较常见,在一般情况下流速可设定为1.5mL/min。[/b][/color][color=#3e3e3e][b](4)有机相的选择[/b]获得成功分离的另一个重要因素就是流动相有机溶剂的选择,如果使用反相色谱柱可以有三种有机溶剂可以选择,甲醇、乙腈和四氢呋喃。每一种溶剂都有其独特的优点,但色谱分析人员很少能预见哪一种溶剂更合适,所以具体选择哪一种溶剂你必须充分考虑同行的意见。分析药物中的成份时,其中有些样品的紫外吸收很弱,所以,分析时紫外检测器的波长为220nm甚至更低,但是四氢呋喃的紫外吸收比较强,所以在波长低于240nm时,就不能选择这种溶剂。尽管低浓度的甲醇在较低的波长下也可使用,但是在低于220nm时,甲醇的浓度不易控制。选择何种溶剂还必须考虑其与样品及空气之间不发生作用,而四氢呋喃易分解,形成过氧化物,所以使用这种溶剂时须格外小心。一些研究人员发现,在四氢呋喃中加入水可以解决这个问题,但它与色谱柱平衡所需的时间比甲醇、乙腈长,而且其不愉快的气味也是一个不利因素。与四氢呋喃相比,甲醇和乙腈的最大优点是在柱压较低的条件下,流速可控制在1-2mL/min之间。考虑到理想溶剂的特点,如粘度低、紫外吸收弱、与样品不相互作用,而且使用方便,所以首选的有机溶剂应是乙腈,当然利用甲醇作为有机溶剂的也较为常见。[b](5)水相的选择[/b]假如样品为一般化合物,可以用水作为水相,然而离子化合物在药物分析时普遍存在,而这类化合物需要控制pH值才能得到很好的分离。如流动相的pH值必须高于或低于样品的PKA1.5个单位。在分析有机酸时,当pH值低于3时,色谱柱一般还比较稳定,然而当PH值高于8时,就需要缓冲液,因为此pH值已经超出了二氧化硅的有效使用范围。宽PH值的二氧化硅柱也比较常见,但是对高pH值物质的分离,很少有这方面的报道。所以建议使用缓冲液来减少二氧化硅的流失。考虑到样品的特性及柱子的稳定,建议在分析PH值较高的物质时应使用缓冲液,2.5mm的磷酸盐缓冲液 (pH=2.5)是非常合适的水相。如果在分析方法中使用了分光仪,那么就必须选择易挥发的缓冲液,尽管不如真正的缓冲液有用,但0.1%Trifluoroacetic酸和乙酸能够满足pH值的控制要[b]求。(6)其它因素[/b]在你选择液相色谱的分析方法时,必须考虑到其它的一些因素,比如,温度。因为温度变化1度,保留时间将变化1-3%,所以温度控制十分重要,温度的变化还影响色谱柱的选择性,这会使你更积极地投入到温度选择中去。在分析时柱温一般比室温高一点(如,35℃),因为此温易控制,而且在低压下有利于降低溶剂的粘度,从而降低柱压。有时你需要利用其它的一些方法 ,如在流动相中加入部分特殊的物质,不过建议最好在你实在必须时才用这种方法,记住KISS原则(Keep it simple ,stupid),不要使你的流动相过分复杂![/color][color=#3e3e3e][b](7)总结[/b]在液相色谱分析时条件的选择非常重要,而且流动相是常年与之打交道的,所以必须慎重选择。一些较为常见的分析条件如表二所示:[/color][color=#3e3e3e][b][img=,529,146]http://ng1.17img.cn/bbsfiles/images/2017/05/201705142214_01_3214098_3.png[/img][/b]表二:分析条件内容[/color][color=#3e3e3e](转载于:实验与分析)[/color]
[align=center]高效液相色谱在中药研究中的应用进展[/align] 液相色谱分析是指流动相为液体的色谱技术,是色谱法中最古老的一种,但通过改进填料的粒度及柱压,在经典的液相柱色谱的基础上引入了[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的塔板理论,在技术上采用了高压输液泵,高效固定相和高灵敏度的检测器,实现了分析速度快、分离效率高和操作自动化,这种色谱技术被称为高效液相色谱法(HighperformanceliquidchromatographyHPLC)。中药的成分非常复杂,以往常用的薄层色谱等方法因其精密度、准确度、灵敏度、重现性差而不能满足现代中药的需要。高效液相色谱正是以其稳定、可靠、高效的特点成为中药研究的最重要的分析方法。目前高效液相色谱已经广泛应用于生物碱、皂苷、黄酮、蒽醌、香豆素等各种中药有效成分的测定。近年来对高效液相色谱监测中药的研究非常多,由于高效液相色谱集经典液相色谱和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的优势于一身,无论柱效、选择性还是分析程度都达到或超过了它们,近年来对高效液相色谱的不足之处进行了改进,使这项技术日臻完善。[b]1 高效液相色谱发展近况 [/b] 高效液相色谱在药物分析中的应用,主要考虑试样的预处理和分析柱、检测器的选择。在试样的预处理上,日前兴起的固相(微)萃取使得许多含量低的成分得到精制提纯,从而适于高效液相色谱的测定,而孙新国采用逆流萃取测定川芎嗪含量取得了很好的效果。中药中有些紫外吸收弱,或无特征紫外吸收的成分,直接用高效液相色谱测定,其灵敏度和分离度都不尽人意,利用柱前或柱后衍生化法可使这些成分较精确地测定出来。 对于极性大、脂溶性差物质,在YWGCl8柱上不易保留,用十二烷基磺酸钠作为离子对试剂,降低其极性,延长柱上的保留时间,取得较好的分离较果。将液相色谱和质谱这两个强有力的分析技术在线连接在一起,经过三十年的发展已成为一项较为成熟的分析手段,但是它从形成伊始就存在着问题:从液相色谱流进质谱时,流动相的变化、溶剂的组成、高温高压离子化的问题制约着这种联用技术发展,大气压离子化接口具有去除溶剂和离子化的双重功效,它的引入,使得该技术在各个领域得到了广泛的应用。电喷雾离子源是一种软电离技术,一般只生成(M+H)+和(M-H)-两种分子离子峰,选择性监测(mz)190的负分子离子峰,具有较高的灵敏度、准确度、专一性,满足了低浓度药物研究的需求。 三维高效液相色谱法可以同步测定葛根素和阿魏酸两种指标。通过实验证明:如果选择合适的柱温等色谱条件,乙醇作为反相高效液相色谱流动相,分析中药及中成药中有效成分,既安全又准确。结构相似的物质,普通的检测器难以检测出来,高效液相色谱-电化学法可以有效地测定黄连粉中仅差一个基团的黄芩苷和黄芩素的含量。样品经色谱柱分离后收集,再经荧光分光光度计测荧光强度,影响因素多,测定复杂,改进后的高效液相色谱-荧光法则可以不经衍生化和收集分离物,只经化学处理除杂,浓缩后直接进样即可。用该法测定贯叶连翘中金丝桃素的含量也取得了较好的结果。高效液相色谱-示差折光测黄芪精口服液中黄芪甲苷的含量也都取得了较为满意的结果。 对于只有紫外末端吸收,用紫外检测时灵敏度低,基线易漂移,示差折光检测其易受外界条件干扰,蒸发散射检测器能克服以上不足,响应值只与样品质量有关,其信号相应与质量成正比,不同化合物,质量相同则信号相应基本一致。蒸发光散射检测法是基于不挥发样品分子对光的散射程度与其质量成正比,与其所含基团性质无关。 只要选择适当的检测器参数,便可使流动相和溶剂完全气化,不产生信号,而样品中的各个组分以不挥发粒子存在,对光有散射,以被检测出来。因此,蒸发光散射检测器可用于含不同基团的多组分同时分离、分析。和紫外、荧光等方法相比,蒸发光散射检测法对不同物质有近似相同的响应因子,因而不出现低浓度、高响应或高浓度、低响应的现象,有利于不同比例混合物的准确测定.高效液相色谱-蒸发光散射检验法测定银杏叶中萜类内酯含量、红参及育精胶囊中人参皂苷Rg1和Re的含量和藤黄中藤黄酸含量都得到了很好的结果。 [b]2 高效液相色谱的研究动向 2.1 缩短分析时间,提高分离效率[/b]。应用先进的检测仪器和方法,对流动相、固定相进行调节或改变,采用梯度洗脱、柱切换技术有望解决这个问题。梯度洗脱的高效液相色谱法,能分析较宽极性范围的样品,较等度洗脱具有很大的优势,但对于成分更复杂、极性范围更宽的中药样品则有些力不从心。多柱高效液相色谱法又称多维高效液相色谱法。除具有梯度洗脱一样的改变流动相浓度的优点外,还可以改变固定相种类、键合度、粒径、柱长、柱径等及流动相种类、浓度等。[b]2.2 进一步向自动化、智能化及联用技术上发展[/b]。液相色谱与质谱联用在国外已成为测定低浓度生物药品中药物及代谢物的首选方法,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]-MS法测定血浆中HIV-1蛋白酶,准确高效,血浆中残留的内源性组份和其他药物不干扰测定,既节省材料又节约时间。已经应用于体液、血浆、血清中的药物分析。中药复方注射液“清开灵”中的胆酸类物的分析采用液相色谱质谱质谱联用,效果理想。高效液相色谱-核磁共振联用在药物分析方面的作用很不错。新近提出的智能多柱高效液相色谱系统利用切换技术的模块式分离性能,把样品分块的切换进不同性质的色谱柱,再用合适的流动相洗脱。全过程采用智能化控制。[b]3 高效液相色谱在中药分析中的应用前景 [/b]中药研究的大趋势是全成分分析,通过对从单味药到复方的不同配伍、煎煮时间等的研究,才能发现中药中化学成分的变化规律,找到中药机理之间的有机联系。中药成分繁多,且各种成分的性质遍布所有极性段、酸碱范围。实现多成分分析的最简单途径即在一根足够长的色谱柱上,采用温和的流动相,在足够久的时间内洗脱。但这与现代分析要求的简便快速相违。通过大量的应用研究表明,高效毛细管电泳在分析中药成分,尤其在分析高极性化学成分方面有较大优势,在分析大量的复方制剂方面显示了较高的能力。 由于毛细管几乎不会出现高效液相色谱分析中常出现的柱床污染现象,而且用过的毛细管柱只需很短的时间进行冲洗后,即可以进行第二个样品的分析,快速高效且分辨率很高。新兴的毛细管电色谱是集高效液相色谱和毛细管电泳优势于一身的一种新型电分离微柱液相色谱技术,它是将高效液相色谱的多种填料微粒移到毛细管中,以样品与固定相间的相互作用作为分离机制,以电渗流为流动相驱动力的色谱过程。最近,一些先进的检测仪器成功的用在了高效液相色谱分析法上,使得高效液相色谱的应用更广泛,并充分利用高效快速、选择性好、灵敏度高等优点,建立更加系统的成分分析方法。通过与质谱联用、梯度洗脱、柱切换技术、配合先进的检测技术,以及与分子生物学、现代分子药理学相结合,必将在中药分析中发挥很大作用。转自《实验与分析》
[color=#00008B][B]该帖为楼主自己整理,帖中所有楼主撰写的内容未经授权不得转载,否则属侵权违法行为![/B][/color]看到很多版友发帖求助讨论准确性的问题,我特意整理了一个关于定量分析误差问题的帖子,希望对大家有所帮助。高效液相色谱定量分析过程可分为样品的前处理、标准品的配制、进样、色谱分离、检测及数据处理等七个步骤。[B]一 误差的主要来源[/B]随着现在市场销售仪器自动化程度的提高,进样、色谱分离、检测及数据处理等实验环节对实验结果产生的误差越来越小,尤其在高效液相色谱定量分析中,实验结果的误差可能主要来源于样品的前处理及标准品的配制。[B]1、样品的前处理 [/B]样品的萃取率是样品前处理时存在的主要问题。当固-液萃取(含柱分离的前处理方法),液-液萃取时,存在萃取率不高且不稳定的问题。尤其在除去蛋白质时,存在变性蛋白质会吸附一些被测组分,导致萃取率降低的情况。通常,萃取率是通过在式样中添加被测成分在萃取的方法评价的。也就是说,被测成分的增加量和液相色谱中的峰面增大成比例关系,通过这种方法可以确定溶液中被测成分的变化趋势。 如果存在萃取率不稳定的问题,就有必要改变萃取的方法,要预先添加内标,然后萃取。这种情况采用的内标,必须与被测物质的化学结构类似,萃取的萃取率才可能相近。如果回收率不但接近100%而且较为稳定,可以证明这种前处理方法较为可靠。在分析中如果能充分考虑以上误差产生的各种原因,才有可能得到精确的分析结果。 [B]2、标准品的配制[/B]本帖中讨论的问题带有普遍性,影响高效液相色谱分析结果准确性的因素较多,仅在标准溶液的配置过程中,可以分为标准物质的称量,溶液的配制和溶液的储存三个环节。 作为标准溶液使用的标准物质其纯度要求很高,应避免使用纯度不符合要求的试剂,作为标准溶液使用的标准物质具有不可替代性,现在市场有销售的HPLC专用的溶剂及各种标准试剂可供实验选择;其次选择与样品浓度要求相适应的天平,应该尽可能使用高精度的天平,这样才能把由于天平使用带来的称量操作误差降至最低。[B]二 消除误差的方法[/B] 要提高分析结果的准确度,必须考虑在分析过程中可能产生的各种误差,采取有效措施,将这些误差减到最小。[B]1、选择合适的分析方法[/B] 各种分析方法的准确度是不同的。化学分析法对高含量组分的测定能获得准确和较满意的结果,相对误差一般在千分之几。而对低含量组分的测定,化学分析法就达不到这个要求。仪器分析法虽然误差较大,但是由于灵敏度高,可以测出低含量组分。在选择分析方法时,一定要根据组分含量及对准确度的要求,在可能条件下选最佳分析方法。[B]2、增加平行测定的次数[/B] 如前所述增加测定次数可以减少随机误差。在一般分析工作中,测定次数为2—4次。如果没有意外误差发生,基本上可以得到比较准确的分析结果。[B]3、消除测定中的系统误差[/B] 消除测定中系统误差可采取以下措施:其一是做空白实验,即在不加试样的情况下,按试样分析规程在同样操作条件下进行的分析。所得结果的数值称为空白值。然后从试样结果中扣除空白值就得到比较可靠的分析结果。其二是注意仪器校正,具有准确体积的和质量的仪器,如滴定管、移液管、容量瓶和分析天平,都应进行校正,以消除仪器不准所引起的系统误差。因为这些测量数据都是参加分析结果计算的。其三是作对照试验,对照试验就是用同样的分析方法在同样的条件下,用标样代替试样进行的平行测定。将对照试验的测定结果与标样的已知含量相比,其比值称为校正系数。 校正系数=标准试样组分的标准含量/标准试样测定的含量 被测试样的组分含量=测得含量×校正系数 综上所述,在分析过程中检查有无系统误差存在,作对照试验是最有效的办法。通过对照试验可以校正测试结果,消除系统误差。[B]4、样品定量分析过程中的误差[/B]样品处理要尽量减少操作者的技术问题带来的误差,样品的稀释次数、稀释工具都是误差的祸根,应尽量减少稀释次数,稀释工具用高准确度的。样品中的干扰组分会直接影响分析的准确度,而且有些组分会损坏柱子。纯化样品的过程尽量少用蒸发至干的步骤(在色谱分析中这一步又是不可少的),正确操作固相柱萃取、纯化小柱使用的步骤,注意提高每一步的回收率,使用内标法也是一个能准确定量的方法。 在手动进样中进样体积至少是样品定量环管体积的3倍,色谱分离程序要使色谱峰的分离度大于1.5,控制流动相、流量、温度等的平稳。流动相的污染都会抬高基线或减少信噪比,分辨率下降,试验条件的变化,如柱退化、不好的流动相等都能引起保留时间变化,引起一个峰或更多的峰不能被鉴别。正确设定仪器参数,选用合理的数据处理参数,用峰面积计算结果比峰高更精确。[B]三 结论 [/B]以上的分析的是我们能尽量控制的误差,还有一些不是操作者所能控制的误差,如被测定组分易分解、组分的含量高低、介质效应等。我们把能控制的误差减小到最低,那你的结果准确度将更高。
[color=#444444]本人目前在做化肥中吡啶含量的检测,但是受条件的限制,只能用液相色谱仪来进行,[/color][color=#444444]求各位高手来指点一下液相分析吡啶的条件和方法。[/color][color=#444444]另外想知道吡啶的浓度达到多少的时候会出现臭味。[/color]
液相色谱检定中,注入低浓度萘甲醇溶液后,会出现一个和萘相当高度的鬼峰,和一个负峰转正峰的峰,最后是萘的峰,这个鬼峰和负峰是怎么来的呢?此外在计算最小检测浓度的公式里,用的噪声是从采集开始到结束时间段里的噪声呢还是需要选取一段比较平缓的基线噪声呢?
1.液相色谱柱的使用说明:(1)液相色谱柱色谱柱使用前注意事项:液相色谱柱的储存液无特殊说明,均为评价报告所示的流动相。在使用前,一定要注意液相色谱柱的储存液与要分析样品的流动相是否互溶。在反相色谱中,如用高浓度的盐或缓冲液作洗脱剂,应先用10%左右的低浓度的有机相洗脱剂过渡一下,否则缓冲液中的盐在高浓度的有机相中很容易析出,堵塞色谱柱。(2)流动相:流动相中所使用的各种有机溶剂要尽可能使用色谱纯,配流动相的水最好是超纯水或全玻璃器皿的双蒸水。如果将所配得流动相再经过0.45μm 的滤膜过滤一次则更好,尤其是含盐的流动相。另外,装流动相的容器和色谱系统中的在线过滤器等装置应该定期清洗或更换。以常规硅胶为基质的键合相填料通常的pH值适用范围是2.0-8.0。当必须要在pH值适用范围的边界条件下使用色谱柱时,每次使用结束后立即用适合于色谱柱储存并与所使用的流动相互溶的溶剂清洗,并完全置换掉原来所使用的流动相。(3)样品:样品也要尽可能清洁,可选用样品过滤器或样品预处理柱(spe)对样品进行预处理;若样品不便处理,要使用保护柱。在用正相色谱法分析样品时,所有的溶剂和样品应严格脱水。2.色谱柱的保存(1)反相液相色谱柱色谱柱每天实验后的保养:使用缓冲液或含盐的流动相,实验完成后应用10%的甲醇/水冲洗30分钟,洗掉色谱柱中的盐,再用甲醇冲洗30分钟。注意:不能用纯水冲洗柱子,应该在水中加入10%的甲醇,防止将填料冲塌陷。(2)长期保存色谱柱:如色谱柱要长时间保存,必须存于合适的溶剂下。对于反相柱可以储存于纯甲醇或乙腈中,正相柱可以储存于严格脱水后的纯正己烷中,离子交换柱可以储存于水(含防腐剂叠氮化钠或柳硫汞)中,并将购买新色谱柱时附送的堵头堵上。储存的温度最好是室温。3.色谱柱在使用过程中易出现的问题和解决办法色谱柱在使用中最常见的问题就是柱压升高,如果柱压是在长时间使用过程中缓慢增加,属于正常现象。但柱压在使用过程中突然升高(系统管路堵塞及压力传感器故障除外),可能的原因有如下几点:(1)色谱柱头的过滤筛板堵塞或污染;(2)色谱柱头的填料被样品污染;(3)色谱柱内缓冲液中的盐遇到高浓度的甲醇或其他有机溶剂,形成结晶析出;(4)流动相pH值过大或过小使固定相结构破坏或溶解。解决办法如下:(1)如确定是色谱柱头的过滤筛板被污染,可以将色谱柱反方向用甲醇冲洗至正常压力,或者卸下色谱柱头,将其放在10%的稀硝酸内超声清洗10分钟,后再用纯水超声10分钟,重新装入色谱柱。(2)如确定色谱柱头的填料被污染,将柱头螺丝卸下,挖出柱内前段被污染的填料,用相同的柱填料重新填入,仔细修复后,重新安装上柱头螺丝。(3)如确定定是盐结晶,用10%的甲醇/水冲洗色谱柱使柱内盐全部溶解,再换高浓度甲醇。(4)如果因pH值使用不当,很难恢复。【来源:实验与分析】
用液相色谱分析废水中酚的含量,进样时酚标液应配成多大浓度?测废水中酚含量时,是直接用废水中酚与酚标液的峰峰面积比计算呢,还是将酚标液配成不同浓度作出标准曲线,从曲线上读呢?谢谢
[color=#444444]对苯二甲酸二异辛酯,间苯二甲酸二异辛酯,邻苯二甲酸二异辛酯液相色谱分析用什么液相色谱柱?[/color]
浅谈高效液相色谱分析中常见问题及对策高效液相色谱(HPLC)作为一种分离技术和方法,目前已经发展到一个全新的阶段。高精度的输液泵,应用广泛的色谱分离柱,低噪音、高灵敏度的各种检测器和功能强大的数据处理软件系统的出现,都推动了液相色谱技术的迅猛发展。液相色谱仪正以它分辨率高、分析速度快和应用广泛的优点倍受仪器分析工作者的青睐,广泛地应用于医药卫生、环境监测、食品检测等领域。作者本人从事液相色谱分析分析工作二十多年,应用HPLC技术在血药浓度、生物胺、核普酸、蛋自质浓度测定等实际工作中,积累了许多的方法和经验,在这里与大家交流,希望能对同行们有所帮助和借鉴,共同促进液相色谱分析技术的发展。1 高效液相色谱仪的基本工作原理 高效液相色谱仪如图1所示,是由溶液贮器、高压泵、进样系统、色谱分离柱、检测器和数据处理系统几部分组成。http://www.came-online.org/userfiles/090109150117044709ny0sff.jpg
液相色谱怎样确定样品的浓度?我先跑出标样的谱图,后跑出样品的谱图,怎样分析得到样品的浓度?如果没有标样能否确定样品的浓度?怎样确定啊?
[align=center]高效液相色谱组分定性分析方法简述[/align]1、保留时间对照法(1)外标对照法 即在相同的色谱条件下,分别进样品溶液与高纯度的单一组分对照品溶液,这里推荐先进样品溶液,确定系统适应性没有问题了再进对照品溶液,对比两组分的保留时间,一般保留时间相对差异在5%以内,同时绝对误差在0.1min以内的认定为同一个物质,但仍遵守“同一组分保留时间肯定一样,但保留时间一样不一定是同一组分,保留时间不一样的肯定不是同一组分”的原则。(2)标准加入法 即在相同的色谱条件下,将高纯度的对照品加入样品溶液中再上机检测,与相同浓度未加对照品的样品溶液比较,峰高或峰面积应呈等比例增加的,可认定为是同一物质。此法比较适用于组分比较复杂,邻近有干扰组分峰时。上述方法尽量使用柱效高或长柱,峰高尽量小一些,甚至可以小到定量限浓度,一些在高响应下是单个峰的,在低响应时可能是两个峰,所以一定要注意峰形,只要峰形与对照品不一致,就要怀疑不是同一个物质。再严谨一点,还可以改变流动相组成、色谱柱、柱温等,样品中的组分峰应与对照品的组分峰有相同的变化。2、利用检测器选择性进行鉴别(1)DAD检测器波长扫描法 对于配有DAD检测器的还可以对比三维扫描图谱和峰纯度计算进行辅助定性。如果没有配DAD检测器,但有具波长扫描功能的可变波长检测器的,可以使用停泵扫描功能,查看扫描图谱进行鉴别。(2)质谱检测器鉴别法 该方法适用于联用有质谱分析仪的高效液相色谱仪,通过比较碎片峰进行比较鉴别。3、破坏法 将物质进行化学破坏,可以是酸、碱降解,也可以是衍生后再进行检测,看组分峰的变化。当然,此方法不适用于组分复杂的样品。
1 高效液相色谱法的特点高效液相色谱法是20世纪70年代急剧发展起来的一项高效、快速的分离分析技术。液相色谱法是指流动相为液体的色谱技术。在经典的液体柱色谱法基础上,引入了气相色谱法的理论,在技术上采用了高压泵、高效固定相和高灵敏度检测器,实现了分析速度快,分离效率高和操作自动化。这种柱色谱技术称做高效液相色谱法。它可用来作液固吸附,液液分配,离子交换和空间排阻色谱(即凝胶渗透色谱)分析,应用非常广泛。高效液相色谱法具有以下几个突出的特点。1.1 高压 液相色谱法以液体作为流动相(称为载液),液体流经色谱柱时,受到的阻力较大,为了能迅速地通过色谱柱,必须对载液施加高压。在现代液相色谱法中供液压力和进样压力都很高,一般可达到150~350×105Pa。高压是高效液相色谱法的一个突出特点。1.2 高速 高效液相色谱法所需的分析时间较之经典液体色谱法少得多,一般都小于1h,例如分离苯的羟基化合物七个组分,只需要1min就可完成;对氨基酸分离,用经典色谱柱,柱长约170㎝、柱径0.9㎝、流动相流速30mL·h-1,需用20多小时才能分离出20种氨基酸,而用高效液相色谱法,只需1h之内即可完成。载液在色谱柱内的流速较之经典液体色谱法高得多,一般可达1~10mL·min-1。1.3高效 气相色谱法的分离效能很高,柱效约为2000塔板/米;而高效液相色谱法的柱效更高,约可达3万塔板/米以上。这是由于近年来研究出了许多新型固定相(如化学键合固定相),使分离效率大大提高。1.4高灵敏度 高效液相色谱已广泛采用高灵敏度的检测器,进一步提高了分析的灵敏度。如紫外检测器的最小检测量可达纳克数量级(10-9g);荧光检测器的灵敏度可达10-11g。高效液相色谱的高灵敏度还表现在所需试样很少,微升数量级的试样就足以进行全分析。高效液相色谱法由于具有上述优点,因而在色谱文献中又将它称为现代液相色谱法,高压液相色谱法或高速液相色谱法。气相色谱法虽具有分析能力好,灵敏度高,分析速度快,操作方便等优点,但是受技术条件的限制,沸点太高的物质或热稳定性差的物质都难于应用气相色谱法进行分析。而高效液相色谱法,只要求试样能制成溶液,而不需要气化,因此不受试样挥发性的限制。对于高沸点、热稳定性差、相对分子质量大(大于400以上)的有机物(这些物质几乎占有机物总数的75%~80%)原则上都可用高效液相色谱法来进行分离、分析。高效液相色谱法的基本概念及理论基础,如保留值、分配系数、分配比、分配度、塔板理论、速率理论等与气相色谱法是一致的,但有其不同之处。液相色谱法与气相色谱法的主要区别可归结于流动相的不同。液相色谱法的流动相为液体,气相色谱法的流动相为气体。液体的扩散系数只有气体的万分之一至十万分之一、液体的粘度比气体大一百倍,而密度为气体的一千倍左右。这些差别显然将对色谱过程产生影响。
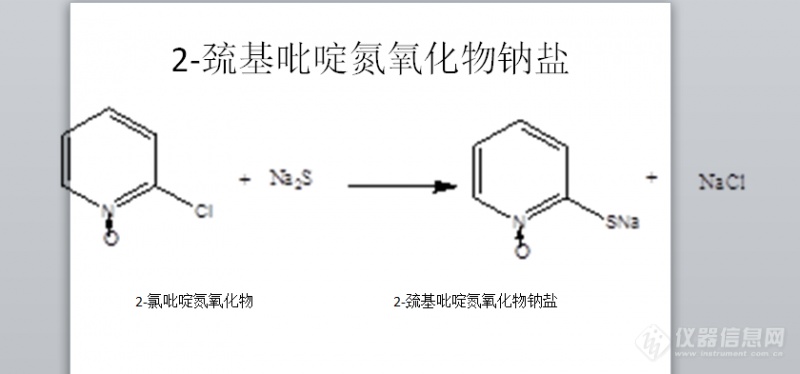
[color=#444444]2-巯基吡啶氮氧化物钠盐怎么用液相分析?[/color][color=#444444]拖尾严重怎么解决?[/color][color=#444444]加磷酸作为缓冲液吗?浓度多少啊[/color][color=#444444]磷酸有液相色谱级的吗[/color][color=#444444][img=,690,322]https://ng1.17img.cn/bbsfiles/images/2019/08/201908071539349672_6238_1849104_3.png!w690x322.jpg[/img][/color]
在测试中心液相色谱组呆了20多年,见过无数的客户,不同的客户对色谱的理解不一。很多人认为色谱就是打一针,出个谱图而已,容易的很,跟他们的科研档次无法比。觉得样品给你了,你必须做出来,而且很快得到其所需要的结果,全然不管你的仪器能否满足要求。做不好或者不想做,就会去告状,服务态度不好之类的。所以一旦征求意见,必然出现各种各样的服务问题。测试人员的地位在学校可见一斑。也有少数本身做液相色谱的,这些人沟通起来非常融洽,因为知道其实际做起来不容易,可惜这类的客户仅有百分之几。在色谱分析的三大分支中,我对液相、离子熟悉,对气相只是很了解,没动手做过。对于这三大类型,其实在实际中做的差别是很大的。先以我精通的离子色谱而言,我几乎涉及了所有的类型,离子色谱能否做关键在于设备,常规的很简单,特殊的全靠设备支撑,因为大部分离子色谱的分析都是优化的方法,固定的模式做起来并不难。但非常规的样品,则难度大大增加,即使同一个组分,由于基体差别,分析方案也是千差万别。也就是常规的很简单,特殊的则很难。现在厂家离子色谱方法开发就是一种特化的过程。离子色谱主要分析离子以及一些极性的化合物,表面上看应用范围比较窄,其实在很多领域有很好的应用,可以解决液相色谱无法解决的一些问题。对于气相色谱,复杂性比离子色谱要高,因为被测的有机化合物种类大大增加,但从气相的结构看,其载气的选择是非常有限的,主要靠色谱柱(极性,非极性,弱极性等),分离则依赖温度的程序升温,它真正的变化在色谱柱,在一般的分析中,大多变化在温度,柱子的变化并不多。因此对于气相色谱,基本就几根柱子。当然一些特别的检测则需要更高级特殊的装置,这跟离子色谱一样。由于受沸点的制约,气相色谱的应用受到很大的限制。而对于液相色谱,就我20年的经历,我认为其复杂性远远高于离子色谱和气相色谱。因为其变化比前二者更多,一是液相色谱分离有很多机理,每种机理都有对应的色谱柱类型,液相色谱的分离机理大约有十来个,很少有人会用过全部机理类型的色谱柱。虽然反相是最常见的分离手段,但由于反相的广泛使用,C18柱的变化类型极多差异很大,这不同C18柱之间的差异有时不亚于不同机理之间的差异。二是,液相色谱最大变化是流动相,不仅有机相类型有变,添加剂类型和浓度有变,不同pH差别很大,面对变化无穷的样品,这个流动相选择变化规律全靠长期的经验积累,很难用文字一言以蔽之。三是,液相色谱的检测器类型最多,离子色谱就三种,气相四五种,而液相色谱的检测器有十来种,相互之间差别极大,不同的检测器对色谱分离机理也有很大的选择性。因此要做好一张液相色谱图,很多情况下只能是你现有条件下的最佳分离,并不是这个化合物的最佳分析条件。对于特殊样品的分析,液相色谱更多的依赖于检测器和柱子的变化,同离子色谱不同。给你一个样品,用那类色谱(液相、气相还是离子),什么柱和条件,则完全依赖你的功底和阅历,当你拥有尽可能多的仪器装备,你才能充分发挥你的能力,依据化合物的特点,样品的特性,选择合适的仪器和配置,做出最佳的色谱图。
[color=#444444]建立液相色谱分析16种PAHs方法,标线浓度分别20mg/L、10mg/L、5mg/L、2mg/L、1mg/L、0.5mg/L,审稿意见说让补充方法检出限数据,这个不是很明白,是不是取一定质量的样品按照整个样品处理流程做下来,看是否达到定量限,然后根据结果,再调整样品量,直至刚好达到检测限,此时的样品量即为方法的检测限。[/color]
何正确使用液相色谱柱(1)使用前注意事项:色谱柱的储存液无特殊说明,均为评价报告所示的流动相。在使用前,一定要注意色谱柱的储存液与要分析样品的流动相是否互溶。在反相色谱中,如用高浓度的盐或缓冲液作洗脱剂,应先用10%左右的低浓度的有机相洗脱剂过渡一下,否则缓冲液中的盐在高浓度的有机相中很容易析出,堵塞色谱柱。(2)流动相:流动相中所使用的各种有机溶剂要尽可能使用色谱纯,配流动相的水最好是超纯水或全玻璃器皿的双蒸水。如果将所配得流动相再经过0.45μm的滤膜过滤一次则更好,尤其是含盐的流动相。另外,装流动相的容器和色谱系统中的在线过滤器等装置应该定期清洗或更换。以常规硅胶为基质的键合相填料通常的PH值适用范围是2.0-8.0,BDS C18适合于碱性化合物,PH值适用范围为2.0-10.0。当必须要在PH值适用范围的边界条件下使用色谱柱时,每次使用结束后立即用适合于色谱柱储存并与所使用的流动相互溶的溶剂清洗,并完全置换掉原来所使用的流动相。(3)样品:样品也要尽可能清洁,可选用样品过滤器或样品预处理柱(SPE)对样品进行预处理;若样品不便处理,要使用保护柱。在用正相色谱法分析样品时,所有的溶剂和样品应严格脱水。
液相色谱分析条件的选择字体: 小 中 大 | 打印 发布: 2010-09-14 14:46:32 来源: 上海禾工科学仪器有限公司 在液相色谱分析过程中,我们经常遇到的问题主要有二种,一种与液相色谱仪器本身因素有关,如液相色谱的阀门、混合器、检测器的光源以及其它的一些硬件设备。出现这类问题后,如果能找出问题根源,解决起来一般很简单,而且这类问题可以通过对仪器的精心维护来避免;而另一类问题则是分析方法本身造成的问题,如出现色谱峰形状不好、峰与峰之间不能分开、基线飘移等等。不幸的是,如果出现这类问题,看起来似乎很明显,但是要找出原因并解决这类问题却非常困难。为了减少出现这一类型的问题,就必须在分析之前,仔细研究并选择一个好的分析方法,有了一个好的分析方法,就很容易获得理想的分离效果,而且在出现问题是也很便于找出原因。要选择一个好的分析方法,就必须对液相色谱分析的一些基本原则要有一个很深的了解。下面是我们实验室对色谱分析人员进行技能培训的一些基本知识。 一:分析方法选择的基本原则 假如你想做一顿丰盛的晚餐,首先必须看一下食谱,然后检查一下你所需的东西是否齐全,如果少了配料还必须去商店购买,这样你才可能做出一顿可口的晚餐。同样进行液相色谱分析时,也必须按照一定的程序进行,首先你必须要有专门的仪器和试剂,然后有目的地选择分析方法,这样你才可能得到好的分析结果,避免走一些弯路。 二:柱子的选择 在液相色谱分析方法中最重要的就是色谱柱的选择,色谱分析人员面对几百种色谱柱,你从何入手呢?我采取的方法就是订阅 LCGC亚太版中RonMajors 的“色谱柱观察”这一专栏,自1984(1)年以来,RonMajors在这一专栏中介绍许多新柱子、色谱柱新技术、色谱柱使用方法等方面的信息。 现在大部分液相色谱分析都是使用反相液相色谱柱,其中以C18 和C8柱最为流行。然而色谱分析并不是流行歌曲,这两种色谱柱之所以运用广泛是因为在大多数情况下,使用这两种柱子都能获得理想的分离效果。尽管一些样品的分离并不是这两种柱子(如表一所示),但[font=







 我要推广仪器
我要推广仪器
 下载APP
下载APP