高效液相色谱法测定黄芪中黄芪甲苷含量的计算方法
求助~液相色谱含量检测的具体计算方法知道的说一下,谢谢!
我没有接触过这个东西,领导也不在,做了个数据,不知道怎么计算。谁能给个液相含量计算公式不?谢谢!
现在药典里有许多使用高效液相法进行含量检测,其中有一些品种所用对照品与待测物质为不同物质.在计算含量时要不要代入对照品与待测物质的分子量.例如:1\甘草中甘草酸的测定,其所用对照品为甘草酸单铵盐,待测物质为甘草酸 2\ 血竭的含量测定:所用对照品为血竭素高氯酸盐,待测特制为血竭素.个人认为,需要进行相应折算.原因为此方法的吸收原理是基于物质结构内的有色官能团,需要以摩尔浓度计算才能客观反应吸收度与官能团的线性原理 若以质量计算,在进行不同物质之间对比计算时,其他不产生吸收支链,其团会对其产影响.
[b]高效液相色谱法的计算方法[/b]高效液相色谱法是用高压输液泵将具有不同极性的单一溶剂或不同比例的混合溶剂、缓冲液等流动相泵入装有固定相的色谱柱,经进样阀注入供试品,由流动相带入柱内,在柱内各成分被分离后,依次进入检测器,色谱信号由记录仪或积分仪记录。 1.对仪器的一般要求 所用的仪器为高效液相色谱仪。色谱柱的填料和流动相的组分应按各品种项下的规定.常用的色谱柱填料有硅胶和化学键合硅胶。后者以十八烷基硅烷键合硅胶最为常用,辛基键合硅胶次之,氰基或氨基键合硅胶也有使用;离子交换填料,用于离子交换色谱;凝胶或玻璃微球等,用于分子排阻色谱等。注样量一般为数微升。除另有规定外,柱温为室温,检测器为紫外吸收检测器。 在用紫外吸收检测器时,所用流动相应符合紫外分光光度法(附录Ⅳ A)项下对溶剂的要求。 正文中各品种项下规定的条件除固定相种类、流动相组分、检测器类型不得任意改变外,其余如色谱柱内径、长度、固定相牌号、载体粒度、流动相流速、混合流动相各组分的比例、柱温、进样量、检测器的灵敏度等,均可适当改变, 以适应具体品种并达到系统适用性试验的要求。一般色谱图约于20分钟内记录完毕。 2.系统适用性试验 按各品种项下要求对仪器进行适用性试验,即用规定的对照品对仪器进行试验和调整,应达到规定的要求;或规定分析状态下色谱柱的最小理论板数、分离度和拖尾因子. (1) 色谱柱的理论板数(n) 在选定的条件下,注入供试品溶液或各品种项下规定的内标物质溶液,记录色谱图,量出供试品主成分或内标物质峰的保留时间t(以分钟或长度计,下同,但应取相同单位)和半高峰宽(W),按n=5.54(t/W)计算色谱柱的理论板数,如果测得理论板数低于各品种项下规定的最小理论板数,应改变色谱柱的某些条件(如柱长、载体性能、色谱柱充填的优劣等),使理论板数达到要求。 (2) 分离度 定量分析时,为便于准确测量,要求定量峰与其他峰或内标峰之间有较好的分离度。分离度(R)的计算公式为: 2(t-t) R= ──W+W 式中 t为相邻两峰中后一峰的保留时间; t为相邻两峰中前一峰的保留时间; W及W为此相邻两峰的峰宽。 除另外有规定外,分离度应大于1.5。 (3) 拖尾因子 为保证测量精度,特别当采用峰高法测量时,应检查待测峰的拖尾因子(T)是否符合各品种项下的规定,或不同浓度进样的校正因子误差是否符合要求。拖尾因子计算公式为: W T=────── 2d 式中 W为0.05峰高处的峰宽; d为峰极大至峰前沿之间的距离。 除另有规定外,T应在0.95~1.05间。 也可按各品种校正因子测定项下,配制相当于80%、100%和120%的对照品溶液,加入规定量的内标溶液,配成三种不同浓度的溶液,分别注样3次,计算平均校正因子,其相对标准偏差应不大于2.0%。 3.测定法 定量测定时,可根据样品的具体情况采用峰面积法或峰高法。但用归一法或内标法测定杂质总量时,须采用峰面积法。 (1) 面积归一化法 测定供试品(或经衍生化处理的供试品)中各杂质及杂质的总量限度采用不加校正因子的峰面积归一法。计算各杂质峰面积及其总和,并求出占总峰面积的百分率。但溶剂峰不计算在内。色谱图的记录时间应根据各品种所含杂质的保留时间决定,除另有规定外,可为该品种项下主成分保留时间的倍数。 (2) 主成分自身对照法 当杂质峰面积与成分峰面积相差悬殊时,采用主成分自身对照法。在测定前,先按各品种项下规定的杂质限度,将供试品稀释成一定浓度的溶液作为对照溶液,进样,调节检测器的灵敏度或进样量,使对照溶液中的主成分色谱峰面积满足准确测量要求。然后取供试品溶液,进样,记录时间,除另有规定外,应为主成分保留时间的倍数。根据测得的供试品溶液的各杂质峰面积及其总和并和对照溶液主成分的峰面积比较,计算杂质限度。 (3) 内标法测定供试品中杂质的总量限度 采用不加校正因子的峰面积法。取供试品,按各品种项下规定的方法配制不含内标物质的供试品溶液,注入仪器,记录色谱图I 再配制含有内标物质的供试品溶液,在同样的条件下注样,记录色谱图Ⅱ。记录的时间除另有规定外,应为该品种项下规定的内标峰保留时间的倍数,色谱图上内标峰高应为记录仪满标度的30%以上,否则应调整注样量或检测器灵敏度。 如果色谱图Ⅰ中没有与色谱图Ⅱ上内标峰保留时间相同的杂质峰,则色谱图Ⅱ中各杂质峰面积之和应小于内标物质峰面积(溶剂峰不计在内)。如果色谱图Ⅰ中有与色谱图Ⅱ上内标物质峰保留时间相同的杂质峰,应将色谱图Ⅱ上的内标物质峰面积减去色谱图Ⅰ中此杂质峰面积,即为内标物质峰的校正面积;色谱图Ⅱ中各杂质峰总面积加色谱图Ⅰ中此杂峰面积,即为各杂质峰的校正总面积,各杂质峰的校正总面积应小于内标物质峰的校正面积。 (4) 内标法加校正因子测定供试品中某个杂质或主成分含量 按各品种项下的规定,精密称(量)取对照品和内标物质,分别配成溶液,精密量取各溶液,配成校正因子测定用的对照溶液,取一定量注入仪器,记录色谱图,测量对照品和内标物质的峰面积或峰高,按下式计算校正因子: A/m 校正因子(f)=─ A/m 式中 A为内标物质的峰面积或峰高;A为对照品的峰面积或峰高; m为加入内标物质的量; m为加入对照品的量。 再取各品种项下含有内标物质的供试品溶液,注入仪器,记录色谱图,测量供试品(或其杂质)峰和内标物质的峰面积或峰高,按下式计算含量:A 含量(m)=f×──A/m 式中 A为供试品(或其杂质)峰面积或峰高;m为供试品(或其杂质)的量; f、A和m的意义同上。 当配制校正因子测定用的对照溶液和含有内标物质的供试品溶液使用同一份内标物质溶液时,则配制内标物质溶液不必精密称(量)取。 (5) 外标法测定供试品中某个杂质或主成分含量,按各品种项下的规定,精密称(量)取对照品和供试品,配制成溶液,分别精密取一定量,注入仪器,记录色谱图,测量对照品和供试品待测成分的峰面积(或峰高),按下式计算含量:A 含量(m)=m×── A 式中各符号意义同上由于微量注射器不易精确控制进样量,当采用外标法测定供试品中某杂质或主成分含量时,以定量环进样为好。来源:分析化学网。[em61]
用液相标准曲线法检测含量时,如果样品面积在标准曲线范围之内,但接近上限,对含量计算有影响?
用液相对化合物含量计算一般有哪些方法,前体是我没有标准样品!
高效液相色谱法的计算方法高效液相色谱法是用高压输液泵将具有不同极性的单一溶剂或不同比例的混合溶剂、缓冲液等流动相泵入装有固定相的色谱柱,经进样阀注入供试品,由流动相带入柱内,在柱内各成分被分离后,依次进入检测器,色谱信号由记录仪或积分仪记录。1.对仪器的一般要求所用的仪器为高效液相色谱仪。色谱柱的填料和流动相的组分应按各品种项下的规定.常用的色谱柱填料有硅胶和化学键合硅胶。后者以十八烷基硅烷键合硅胶最为常用,辛基键合硅胶次之,氰基或氨基键合硅胶也有使用;离子交换填料,用于离子交换色谱;凝胶或玻璃微球等,用于分子排阻色谱等。注样量一般为数微升。除另有规定外,柱温为室温,检测器为紫外吸收检测器。在用紫外吸收检测器时,所用流动相应符合紫外分光光度法(附录Ⅳ A)项下对溶剂的要求。正文中各品种项下规定的条件除固定相种类、流动相组分、检测器类型不得任意改变外,其余如色谱柱内径、长度、固定相牌号、载体粒度、流动相流速、混合流动相各组分的比例、柱温、进样量、检测器的灵敏度等,均可适当改变,以适应具体品种并达到系统适用性试验的要求。一般色谱图约于20分钟内记录完毕。2.系统适用性试验按各品种项下要求对仪器进行适用性试验,即用规定的对照品对仪器进行试验和调整,应达到规定的要求;或规定分析状态下色谱柱的最小理论板数、分离度和拖尾因子.(1) 色谱柱的理论板数(n)在选定的条件下,注入供试品溶液或各品种项下规定的内标物质溶液,记录色谱图,量出供试品主成分或内标物质峰的保留时间t(以分钟或长度计,下同,但应取相同单位)和半高峰宽(W),按n=5.54(t/W)计算色谱柱的理论板数,如果测得理论板数低于各品种项下规定的最小理论板数,应改变色谱柱的某些条件(如柱长、载体性能、色谱柱充填的优劣等),使理论板数达到要求。(2) 分离度定量分析时,为便于准确测量,要求定量峰与其他峰或内标峰之间有较好的分离度。分离度(R)的计算公式为: 2(t-t) R= ──W+W 式中 t为相邻两峰中后一峰的保留时间; t为相邻两峰中前一峰的保留时间; W及W为此相邻两峰的峰宽。 除另外有规定外,分离度应大于1.5。(3) 拖尾因子 为保证测量精度,特别当采用峰高法测量时,应检查待测峰的拖尾因子(T)是否符合各品种项下的规定,或不同浓度进样的校正因子误差是否符合要求。拖尾因子计算公式为: W T=────── 2d 式中 W为0.05峰高处的峰宽; d为峰极大至峰前沿之间的距离。 除另有规定外,T应在0.95~1.05间。 也可按各品种校正因子测定项下,配制相当于80%、100%和120%的对照品溶液,加入规定量的内标溶液,配成三种不同浓度的溶液,分别注样3次,计算平均校正因子,其相对标准偏差应不大于2.0%。3.测定法 定量测定时,可根据样品的具体情况采用峰面积法或峰高法。但用归一法或内标法测定杂质总量时,须采用峰面积法。 (1) 面积归一化法 测定供试品(或经衍生化处理的供试品)中各杂质及杂质的总量限度采用不加校正因子的峰面积归一法。计算各杂质峰面积及其总和,并求出占总峰面积的百分率。但溶剂峰不计算在内。色谱图的记录时间应根据各品种所含杂质的保留时间决定,除另有规定外,可为该品种项下主成分保留时间的倍数。(2) 主成分自身对照法当杂质峰面积与成分峰面积相差悬殊时,采用主成分自身对照法。在测定前,先按各品种项下规定的杂质限度,将供试品稀释成一定浓度的溶液作为对照溶液,进样,调节检测器的灵敏度或进样量,使对照溶液中的主成分色谱峰面积满足准确测量要求。然后取供试品溶液,进样,记录时间,除另有规定外,应为主成分保留时间的倍数。根据测得的供试品溶液的各杂质峰面积及其总和并和对照溶液主成分的峰面积比较,计算杂质限度。(3) 内标法测定供试品中杂质的总量限度采用不加校正因子的峰面积法。取供试品,按各品种项下规定的方法配制不含内标物质的供试品溶液,注入仪器,记录色谱图I 再配制含有内标物质的供试品溶液,在同样的条件下注样,记录色谱图Ⅱ。记录的时间除另有规定外,应为该品种项下规定的内标峰保留时间的倍数,色谱图上内标峰高应为记录仪满标度的30%以上,否则应调整注样量或检测器灵敏度。如果色谱图Ⅰ中没有与色谱图Ⅱ上内标峰保留时间相同的杂质峰,则色谱图Ⅱ中各杂质峰面积之和应小于内标物质峰面积(溶剂峰不计在内)。如果色谱图Ⅰ中有与色谱图Ⅱ上内标物质峰保留时间相同的杂质峰,应将色谱图Ⅱ上的内标物质峰面积减去色谱图Ⅰ中此杂质峰面积,即为内标物质峰的校正面积;色谱图Ⅱ中各杂质峰总面积加色谱图Ⅰ中此杂峰面积,即为各杂质峰的校正总面积,各杂质峰的校正总面积应小于内标物质峰的校正面积。(4) 内标法加校正因子测定供试品中某个杂质或主成分含量按各品种项下的规定,精密称(量)取对照品和内标物质,分别配成溶液,精密量取各溶液,配成校正因子测定用的对照溶液,取一定量注入仪器,记录色谱图,测量对照品和内标物质的峰面积或峰高,按下式计算校正因子: A/m 校正因子(f)=─ A/m 式中 A为内标物质的峰面积或峰高;A为对照品的峰面积或峰高; m为加入内标物质的量; m为加入对照品的量。再取各品种项下含有内标物质的供试品溶液,注入仪器,记录色谱图,测量供试品(或其杂质)峰和内标物质的峰面积或峰高,按下式计算含量:A 含量(m)=f×──A/m 式中 A为供试品(或其杂质)峰面积或峰高;m为供试品(或其杂质)的量; f、A和m的意义同上。当配制校正因子测定用的对照溶液和含有内标物质的供试品溶液使用同一份内标物质溶液时,则配制内标物质溶液不必精密称(量)取。(5) 外标法测定供试品中某个杂质或主成分含量,按各品种项下的规定,精密称(量)取对照品和供试品,配制成溶液,分别精密取一定量,注入仪器,记录色谱图,测量对照品和供试品待测成分的峰面积(或峰高),按下式计算含量:A 含量(m)=m×── A 式中各符号意义同上由于微量注射器不易精确控制进样量,当采用外标法测定供试品中某杂质或主成分含量时,以定量环进样为好。来源:分析化学网。
ASTM D790中定义了三种模量:正切模量,割线模量,弦模量。我了解这三者的定义,但想知道它们的适用范围,在什么情况下采用哪种计算方法?另外在正切模量的计算方法中,E=L^3m/4bd^3,其中m为弯曲时切线与负载-弯曲初始直线部分的倾斜度,N/mm,这个m是怎么取的呢?大家在计算过程中是怎样取模量计算点的呢?比如弦模量的两点,比如割线的一点?
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的回收率和液相色谱的回收率在实验设置和计算方法上,有什么不同?
[color=#444444]液相色谱,在不进对照品情况下,如何根据液相图谱面积百分比计算出目标峰的含量?[/color]
大家好,我在用高效液相色谱测芒果苷含量,具体步骤是:取干燥叶片0.1g,加25ML甲醇(萃取剂),称重,超声提取后冷却,用甲醇补足失质量。旋转蒸发仪加热回流,蒸干物用甲醇溶解(这一步是补足失掉甲醇量还是定容到25ML?),最后过滤,滤液体积v1,统一取滤液10ML进行液相色谱测。我只知道液相色谱可以测出芒果苷浓度,但是回归到叶片怎么计算芒果苷含量?哪些体积量是需要用到的。研一新手,实在是搞不懂,恳请大家帮助,谢谢!
今天通过GC-MS数据处理软件建立了塑化剂的定量计算方法,然后用建立好的定量计算方法反过来计算标准溶液中不同塑化剂的含量,发现计算结果比标准溶液的含量少很多,比如,标准1#样品中DMP的实际标准含量是0.05ug/mL, 而通过定量计算方法计算结果只有0.03ug/mL,两者相差太大了,其他成分的塑化剂也是同样的现象,这是为什么呢?ps:DMP标准曲线的R2=1,其他成分的R2也是0.998—1。2. 计算一个样品中塑化剂含量时,发现其积分明显不对,手动积分后,用定量计算方法来计算,发现不管用(即手动积分结果无效一样),还是按照仪器不正确的积分结果进行计算的,这是为什么呢?
液相含量不是要称俩个对照品吗,在计算时是先算出俩个(对照品浓度/峰面积)的平均值,再带入公式计算?还是用每个对照的峰面积直接带入公式计算?
当供试品进样体积是对照品进样体积的2倍,为什么计算含量结果时要在公式后面除以2
在中成药含量计算中,是不是只要成品有水分要求的在含量计算过程中都要扣除水分计算?没说以干燥品计算的!!!
(一)建设工地起尘量计算二)道路起尘量计算(三)一年中单位长度道路的起尘量计算(四)煤堆起尘量计算(五)煤炭装卸起尘(六)汽车道路扬尘[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=188815]起尘计算.rar[/url]
你好,我公司是一个制药公司,样品含量计算之前是用人手动计算,现在打算用软件自动计算,请问这样应该要怎么验证,验证依据哪里能找到
已知总含量为96%标品在HPLC-UV色谱图上有三个峰(买不到单标),峰面积与标品浓度的关系见下表。求峰面积与总浓度的关系,进而计算未知样品的总含量。 [table][tr][td=1,1,72] c[/td][td=1,1,72] A1[/td][td=1,1,72] A2[/td][td=1,1,72] A3[/td][/tr][tr][td=1,1,72] 2.0001[/td][td=1,1,72] 768.7[/td][td=1,1,72] 74.9[/td][td=1,1,72] 691.2[/td][/tr][tr][td=1,1,72] 4.0002[/td][td=1,1,72] 1551.1[/td][td=1,1,72] 148.2[/td][td=1,1,72] 1394.5[/td][/tr][tr][td=1,1,72] 8.0004[/td][td=1,1,72] 3113.3[/td][td=1,1,72] 319.1[/td][td=1,1,72] 2762.2[/td][/tr][tr][td=1,1,72] 40.002[/td][td=1,1,72] 15450.1[/td][td=1,1,72] 1549.8[/td][td=1,1,72] 13736.7[/td][/tr][/table]对组分[i]i[/i], 其峰面积与其浓度的关系如式(1)[img=,91,16]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181706237212_4575_3037435_3.png[/img](1)其中,A峰面积;[i]C[sub]i[/sub][/i] 组分i在样品中的浓度 [i]a[/i], 总含量[align=center] [/align][i]i[/i]个组分的总浓度可表示为式(2),[align=center][img=,102,22]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181706238876_7732_3037435_3.png[/img] (2)[/align]式(1)(2)合并为式(3),即总浓度*总含量是峰面积的函数[img=,150,35]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181706240419_6223_3037435_3.png[/img]方便起见,将式(3)变形,得到多元线性方程(4)[img=,208,20]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181706244891_9083_3037435_3.png[/img]测量四组C,可得到相应的A1,A2,A3解方程或多元最小二乘法回归得到x,y,z,B。 代入未知样品的峰面积和浓度就能得到其总含量。 用mathlab[b]解方程得到x,y,z,B. 但得到了负值解[/b] 这不合常理,x,y,z分别对应斜率的倒数,有物理意义。SPSS最小二乘法回归会剔除两个峰,模型也有问题。请问大神怎么求解?
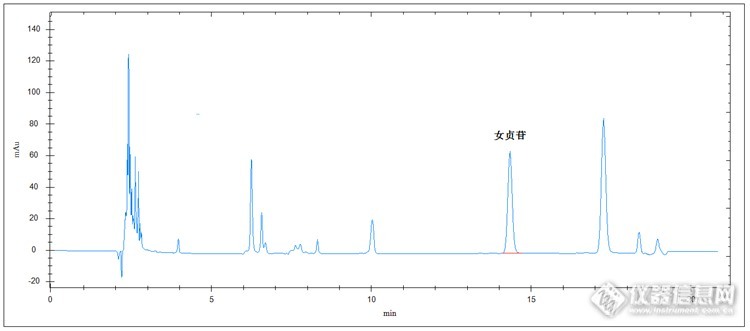
高效液相色谱法检测女贞子药材中女贞苷含量 药材女贞子为木樨科植物,具有滋肝肾、明目黑发,也可治疗眩晕耳鸣、腰膝酸软等症状。其中女贞苷是女贞子中重要成分之一。相应饮片和药材都要求检测女贞苷含量。 以下的方法采用的是高效液相色谱法,检测的是女贞子药材中女贞苷含量。实验部分原理 取适量样品粉碎、溶解、超声提取、定容、滤过,由进样系统进样,色谱柱分离,紫外检测器检测,保留时间定性,峰面积定量计算。仪器及试剂 仪器:高效液相色谱仪(紫外检测器),超声波清洗仪,粉碎设备,三号筛,电子天平,具塞锥形瓶,溶解过滤器,针筒式过滤器 试剂:甲醇(色谱纯),超纯水,稀乙醇样品制备 对照品溶液的制备:精密称取女贞苷对照品12.5mg于50ml容量瓶中,加甲醇制成0.25mg/ml女贞苷溶液,备用。 供试品溶液的制备:取女贞子药材样品适量,充分粉碎后过三号筛,准确称取过筛后的粉末0.5g,置于具塞锥形瓶中,精密加入稀乙醇50ml,称定重量,超声波超声30min,待样品完全溶解后,放冷,再称定重量,用稀乙醇补充减少的重量,摇匀,微膜滤过,待测。色谱条件检测器:紫外检测器色谱柱:C18,4.6 X 250mm,5um波长:224nm流动相:甲醇:水=40:60(V:V)流速:1.0ml/min柱温:30℃进样量:5ul对照品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410191706_518997_2498430_3.png供试品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410191706_518998_2498430_3.png 看看这效果,很羡慕吧,峰形、分离度都很完美吧。 该方法检测女贞子药材中女贞苷含量操作简单、方便,实验快速,结果准确、可靠,是一种坚持女贞子的好方法。 注意的是样品前处理时如果取样量较大或样品粉碎的不够充分,样品提取时可能不能提取完全。那样的话一是样品量要适中,不要太多;二是样品粉碎的要尽量充分;三是超声波提取时间要适当长一些,或提取时温度湿度高一点,或分成几次或几批提取等。 另外这个样品杂质和目标物干扰物都较少,检测时可以适当考虑下缩短检测时间的问题,比如适当增加流动相中甲醇含量,适当升高色谱柱温度控制等。下面我们就看看流动相甲醇:水=50:50,柱温40℃的效果吧。对照品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410191706_518999_2498430_3.png供试品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410191706_519000_2498430_3.png 效果很好吧,这种方法我们在检测时是不是可以考虑下。哈哈,经验之谈,祝大家工作顺利,天天开心!
请问你们高效液相含量的计算公式都有哪些啊?中国心 中国心 中国心
[b]分光光度法原始记录表中包含“[/b]相对含量计算修正值“项,此值如何得到?如何使用?
求氯化钠溶液的含量计算公式,谢谢了~~~真的很急
液相色谱,有结晶水的原料药怎样计算含量我们液相色谱测样品的含量,样品中含两个结晶水,但所用对照品是无水的,计算样品含量时要扣除那两分子的水分吗?

高效液相色谱法检测中药材栀子中栀子苷含量 《栀子花开》,多么动听的歌声,打动多少人的心怀。《栀子花开》,多么精彩的剧情,吸引着多少人的情哀。 大家都知道栀子花很美丽,却可能不知道栀子的果实还是中药材,还有很多药物功效。http://ng1.17img.cn/bbsfiles/images/2015/08/201508262335_563082_2536753_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/08/201508262335_563083_2536753_3.png 栀子果实气微,味微酸而苦,是传统中药,属卫生部颁布的第l批药食两用资源。具有泻火、护肝、利胆、清热、除烦、凉血、降压、镇静、止痛、解毒、止血、消肿、消炎等多种药效。在中医临床常用于治疗黄疸型肝炎、扭挫伤、热病心烦,淋证涩痛,血热吐衄,目赤肿痛,火毒疮疡高血压、糖尿病等病症。 栀子中的栀子苷是赋予栀子药物疗效的主要成分,栀子苷的含量从某种意义上决定着栀子品质和价值,检测必不可少。下面我们就迫不及待的介绍高效液相色谱法检测中药材栀子中栀子苷含量。【实验部分】【原理】 取适量栀子样品经甲醇溶解,超声提取后注入高效液相色谱系统,C18色谱柱分离,紫外检测器检测,外标法(保留时间定性,峰面积定量)计算,得出栀子中栀子苷含量。【仪器及试剂】 仪器:高效液相色谱仪(紫外检测器+高压泵+柱温箱等),超声波清洗仪,溶剂过滤器,电子天平(0.001),四号筛(药典筛)等。 试剂:甲醇(色谱纯),乙腈(色谱纯),超纯水等。【样品溶液制备】 对照品溶液制备:精密称取栀子苷对照品1.5mg于50ml容量瓶中,加甲醇溶解定容,制成30μg/ml栀子苷对照品溶液,备用。 供试品溶液制备:取样品栀子成熟、干燥果实适量,充分粉碎后过药典筛四号筛,精密称取过筛粉末0.1g置于具塞锥形瓶中,精密加入甲醇50ml,称定重量,超声处理20分钟后,放冷,再次称定重量,用甲醇补足减少的重量,摇匀,0.45μm有机相微膜滤过,待测。【色谱条件】检测器:紫外检测器检测波长:238nm色谱柱:Promosil C18 ( 250 mm X 4.6mm,5μm )流动相:乙腈:水=15:85(V:V)流速:1.0ml/min柱温:30℃进样量:10μl【色谱图】对照品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2015/08/201508262336_563084_2536753_3.png供试品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2015/08/201508262336_563085_2536753_3.png【计算】样品中被测物含量计算公式:http://ng1.17img.cn/bbsfiles/images/2015/08/201508262336_563086_2536753_3.pngD----供试品中被测物百分含量,以(%)表示V----供试品稀释的总体积,单位(mL)P3----供试品中被测物峰面积值P4-----对照品中被测物峰面积值ρ2----对照品被测物浓度。单位为mg/mlm2----样品质量,单位(mg)http://ng1.17img.cn/bbsfiles/images/2015/08/201508262336_563087_2536753_3.png【结果】 通过以上公式计算,栀子中栀子苷的含量近似为4.48%,远高于药典要求的不小于1.8%,该栀子中栀子苷含量满足药典要求,该项目检测合格。【结论】 以上方法检测栀子中栀子苷含量方便、快捷,结果准确、可靠,操作简单、易行,该方法适合该类样品检测。
各位老师!用高效液相色谱仪检测纺织品邻苯二甲酸酯如何计算其含量?
有内标物时高效液相色谱法中如何计算含量
药典液相色谱方法的调整 • 根据药典附录“液相色谱法”规定,可调整适当参数 ---调整目的:满足系统适用性的要求 ---系统适用性的要求 ---HPLC方法调整的考虑因素 05版药典的系统适用性要求1、理论塔板数: ----反映整个色谱系统的状态 填料状态 管线连接 ----有不同的计算方法 主要是峰宽取值方法不同 不同计算方法计算结果有差异 ----影响因素: 被测组分的保留时间、进样量等 积分参数 系统死体积 测定色谱方法、样品与计算方法保持恒定,以便比较 05版药典的系统适用性要求 2、分离度: ---影响因素: * 影响柱效的因素 色谱柱尺寸 填料性能 进样量 * 影响分离选择性的因素 流动相组成 色谱柱品牌 柱温 * 柱外体积 ---有不同的计算方法,结果有差异 05版药典的系统适用性要求3、重复性(进样精密度): * 外标法:对照品溶液(n:5) 峰面积RSD:2.0% * 内标法:相当于80%,100%,120%的对照品溶液,加入规定量内标 溶液,分别至少进样2次,计算平均校正因子([font=Times New Roman
各位师傅!用高效液相色谱仪检测纺织品邻苯二甲酸酯如何计算含量。
药典液相色谱方法的调整 • 根据药典附录“液相色谱法”规定,可调整适当参数 ---调整目的:满足系统适用性的要求 ---系统适用性的要求 ---HPLC方法调整的考虑因素 05版药典的系统适用性要求1、理论塔板数: ----反映整个色谱系统的状态 填料状态 管线连接 ----有不同的计算方法 主要是峰宽取值方法不同 不同计算方法计算结果有差异 ----影响因素: 被测组分的保留时间、进样量等 积分参数 系统死体积 测定色谱方法、样品与计算方法保持恒定,以便比较 05版药典的系统适用性要求 2、分离度: ---影响因素: * 影响柱效的因素 色谱柱尺寸 填料性能 进样量 * 影响分离选择性的因素 流动相组成 色谱柱品牌 柱温 * 柱外体积 ---有不同的计算方法,结果有差异 05版药典的系统适用性要求3、重复性(进样精密度): * 外标法:对照品溶液(n:5) 峰面积RSD:2.0% * 内标法:相当于80%,100%,120%的对照品溶液,加入规定量内标 溶液,分别至少进样2次,计算平均校正因子([font=Times New Roman







 我要推广仪器
我要推广仪器
 下载APP
下载APP