请教:液相色谱测定原料药杂质如果结果分别在5%、2%、1%、0.5%、0.1%以内平行结果的相对误差分别应在多少以内?如果是不同仪器、不同色谱柱、同样方法相对误差又该是多少呢?
标准品是异黄酮的4种组分,用的流动相是甲醇:水:醋酸=45:54:1,第一和第二种组分的峰型很好,但是第三和第四种组分的峰总是与杂质峰分不开。我也调整了流动相的比例,进样量也调整过了,但是问题依然存在。(我用的液相色谱仪比较老,只能进行等梯度洗脱)。我是新手,对液相色谱了解的也不是很多,现在很着急啊,连标准曲线都做不好。更别说将来要测量样品中的含量了。
如题。做液相色谱质谱杂质峰丰度很高,达到2*E5,怎么冲都冲不掉,做的是蛋白酶解实验。各位有办法吗?又遇到类似情况吗?
有没有做过维生素A杂质的液相色谱分析?维生素A及其杂质维生素A环氧化物、维生素A醛、维生素A酸没有杂质对照品,以上物质在甲醇-水(90:10)流动相,C18*250mm柱能不能分的开?
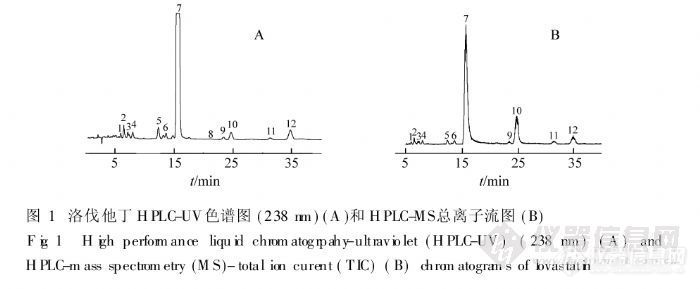
【作者】 吴永江; 朱炜; 邵青; 程翼宇;【Author】 Wu Yongjiang,Zhu Wei,Shao Qin,Cheng Yiyu~*(College of Pharmaceutical Sciences,Zhejiang University,Hangzhou 310031)【机构】 浙江大学药学院; 浙江大学药学院 杭州310031; 杭州310031;【摘要】 利用高效液相色谱-二极管阵列检测器-质谱联用方法对洛伐他丁及其杂质成分进行分离分析和结构鉴定。实验采用D iamonsil C18(5μm,4.6 mm×250 mm)为分离柱,乙腈-水(含0.1%乙酸)(65∶35)为流动相,分离并检测了洛伐他丁及其杂质;通过与DAD检测器和离子阱质谱联用,获得了它们的紫外光谱和质谱数据;紫外光谱表明除氢化洛伐他丁外其余杂质与洛伐他丁基本结构相同,利用MS和MS2数据确定了杂质的分子量和侧链结构,由此鉴定了其中10个杂质的结构。实验结果表明,高效液相色谱-二极管阵列检测器-质谱联用技术可以快速鉴定洛伐他丁中的杂质化学成分。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207301723_380642_2379123_3.jpg
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=102921]多菌灵原药中2个酚嗪类杂质含量的高效液相色谱法测定[/url]
在液相色谱中产品峰哈杂质峰分不开,怎么办?有哪些因素影响分离度?
我用的是日立D-2000高效液相色谱仪器 新的 新的waters 氨基柱子 用错了流动相是 乙二胺四乙酸而那钙盐 ,用的时间很短,后来用水冲洗了很久,又用了乙腈。每次出现的是杂质峰,一大一小。太强了信号(700左右),主峰都看不到啊,杂质峰出峰时间固定,面积固定。。现在怎么办啊。。进什么标样出来的杂质峰都一样啊。。一样看不到主峰在哪儿。进样针进行了多次的清洗。进了多次的乙腈都是倒峰。
高效液相色谱法测定有关物质的方法建立的过程 有关物质系药品中除主成分以外的杂质,有关物质来源的两种途径:1.起始原料及合成过程中的中间体和副产物;2.储存过程中产生的降解产物。 由于有关物质的含量较少,所以选择专属性强、灵敏度高、重现性好的检测方法至关重要。目前常用的方法是高效液相色谱法。现主要阐述高效液相色谱法的建立过程。 高效液相色谱法检测有关物质的方法有:(1)杂质对照品法(适用于已知杂质);(2)主成分自身对照法(紫外检测器),采用HPLC主成分自身对照法检查有关物质,其前提之一是需检查的杂质与主成分在确定的检测波长下应有相近的紫外吸收(响应值接近)。一、色谱条件的确定 1、查阅文献,看看是相关的资料,有的话可以参考。 2、没有文献,进行以下工作。 1)分析结构,看看有无紫外吸收,有就选用紫外检测器,没有的话可以选用蒸发光散射检测器等。 2)分析结构,看看选用何种色谱方式进行分离(正相或者反相色谱)。 3)分析结构,看看流动相该如何选择,要不要选用一些离子对试剂。 4)与同类产品进行比较。 3、紫外扫描一下,找到最大吸收波长。将能得到的中间体、副产物、分解产物、样品配成相同浓度,在紫外扫描分光光度计上扫描一下,选择工艺中最容易带入或产生的杂质的最大吸收处的波长作为测定波长。要是有二极管阵列检测器就不需要这一步了。 4、查找资料,看看类似的样品的流动相,以它为起始流动相进行选择,如果没有,那就找专业书,看看它是怎么选择流动相的。 5、通过专属性验证所选色谱条件能否将各杂质与被测物分离检出 。应按0.5%(w/w)被测物浓度的各杂质量添加至被测物中,模拟被测物中可能存在杂质的状态,即有少量(约0.5%)杂质存在时能否与被测物达到完全分离(分离度大于2.0),以验证系统适用性。二、最低检测限 得出达到S/N=2或S/N=3时的样品药品浓度。三、供试品溶液浓度 一般在样品不过载的情况下,适当设定得高些,以保证该浓度在任何试验条件下,均有足够的检测灵敏度;也可以根据峰面积确定(经验),面积七位数时9开头,八位数时1开头。四、线性及精密度试验 按药典规定,即采用该杂质对照品 (外购或合成获得)以外标法准确测定。此时,与含量测定相似,应进行线性试验。通常将杂质线性验证范围0.05%~1.2%)。精密度试验是以杂质百分浓度为0.5%进样,相对标准偏差不大于2.0。五、回收率试验 杂质的定量试验可向原料药或制剂中加入已知量杂质进行测定。将各杂质以0.4%、0.5%、0.6% (w/w)的浓度加至被测物溶液中,以验证所采用的色谱条件是否可分离检测相应的各杂质以及与被测物中已存在的杂质是否累加,并观测累加量的准确性。六、强降解试验 强降解试验条件有酸、碱、氧化、强光、高温破坏等。破坏时不能过于剧烈,一般以产生10%~20%杂质的条件为宜(《化学药物杂质研究技术指导原则》,同时采用二极管阵列检测器(DAD)验证主峰峰纯度,验证物料平衡。七、其他色谱参数的确定 流速的选择:主峰保留时间约在l0min左右,这样主峰不易出现拖尾、堆积的现象,运行时间也不会太长。柱温的选择:不应超过35℃,既可增强被测物与杂质的分离度,也可避免因温度过高使主成分降解,导致测定误差。 溶剂的选择:首选流动相作溶剂为宜,以排除溶剂峰的干扰。也可以先用和流动相同比例有机溶剂溶解,再加入同比例水相。难溶物质,可采用甲醇或乙腈作溶剂以提高溶解性。 色谱图记录时间的设定:根据强力破坏试验和样品稳定性试验来规定色谱图记录时间,应能洗脱出有可能存在的全部杂质和经强力破坏试验产生的杂质,并规定运行时间主成分保留时间的几倍。八、杂质限度的确定 杂质限度的制订应考虑如下因素:杂质及含一定限量杂质的药品的毒理学研究结果;给药途径;每日剂量;给药人群;杂质药理学可能的研究结果;原料药的来源;治疗周期;在保证安全有效的前提下,药品生产企业对生产高质量药品所需成本和消费者对药品价格的承受力。严格按照《化学药物杂质研究技术指导原则》来制定。
用液相色谱检测控制杂质限量,要求是杂质含量超过0.1%的不能超过三个。但是设定不同的积分参数(峰高,峰面积,峰宽,斜率等),得到的杂质峰个数可能就不一样,请问大家在遇到这种情况的时候是怎么做的?对此有没有标准文件可以参考的?还是全凭经验来?个人有个想法就是根据仪器检测限,即3倍信噪比来确定峰高这一参数,不知道合理不合理?请个人大神指教!
[color=#444444]最近在做一个酯类的液相分析,它在高温条件下容易发生聚合反应,高温降解的样品在现在的液相条件下2min左右就有很多杂质峰出现,但完全不能分离,我们调节了流动相、柱温、流速等都没有效果,请问这种情况应该怎么办,一般用什么方法或者什么柱子来分离比较好[/color]
高效液相色谱仪总有一个杂质峰!以前没有的,突然就有了,现在跑溶剂的时候也有,换了色谱柱还是有。求解答?
如题,液相色谱中,主成分在该杂质的最大吸收波长下是有吸收的,该杂质在此条件下出峰都正常。改换流动相之后,主成分出峰。如果在原条件下进行分析会对该杂质的检测结果有何影响吗?主成分一定要出峰吗?
用液相色谱测定水中的菲,需要什么前处理,样品中有机物杂质含量低,只需定量.用什么检测器,我是菜鸟,请高手指点!谢谢![em0808][em0808]

CNW Athena C18-WP分析原料药及其杂质实验目的我们研究的样品为3.1类样品,其API包括一个位置异构体,其极性非常相近,当有250*4.6,5u C18色谱柱是,其分离度达不到药典标准,并且其流动相水相PH值为2.5±0.1之间,要求其耐酸性,为了更好的分离两物质并且耐酸的柱子,购买了Athena C18-WP。实验方法及结果分析:对色谱柱进行了试验考察:1、色谱条件:C18 250mm×4.6mm,3μm 柱温45℃,以磷酸水溶液(去离子水用磷酸调pH至2.5)为流动相A,以乙腈为流动相B,按下表程序进行梯度洗脱:时间(分)流动相A(%)流动相B(%)0.01604060109060.01Stop/流速:1.0ml/min;检测波长:260nm ;进样体积:10ul2、仪器与试剂:高效液相色谱仪、电子天平色谱纯乙腈、分析纯磷酸、异构体对照品、原料3、线性溶液配制取异构体和原料适量,用乙腈溶解并稀释制得各成分浓度均为0.3μg/ml、0.6μg/ml、0.8μg/ml、1.0μg/ml、1.2μg/ml
高效液相色谱法测定有关物质的方法建立的过程 有关物质系药品中除主成分以外的杂质,有关物质来源的两种途径:1.合成过程中的中间体和副产物;2.储存过程中产生的降解产物。 由于有关物质的含量较少,所以选择专属性强、灵敏度高、重现性好的检测方法至关重要。目前常用的方法有HPLC法和TLC法,后法不讨论。现主要阐述HPLC法的建立过程。 HPLC法检测有关物质的方法有:(1)杂质对照品法(适用于已知杂质);(2)主成分自身稀释对照法(适用于一般杂质检查,杂质成分少且尚不能取得其对照品,简称“自身对照法”);(3)归一化法 (现已不多用)。前法为外标法、定量检测;后两法均为限量检测。一、色谱条件的确定 1、查阅文献,看看是否同行已经做过类似的工作,有的话可以参考。 2、没有文献,进行以下工作。 1)分析结构,看看有无紫外吸收,有就比较简单,选用紫外检测器就行了,没有的话那就麻烦,可以选用蒸发光散射检测器等。 2)分析结构,看看选用何种色谱方式进行分离(正相或者反相色谱)。 3)分析结构,看看流动相该如何选择,要不要选用一些离子对试剂。 4)与同类产品进行比较。 3、紫外扫描一下,找到最大吸收波长。将能得到的中间体、副产物、分解产物、样品配成相同浓度,在紫外扫描分光光度计上扫描一下,选择所有物质具有相同的最大吸收处的波长作为测定波长。要是有二极管阵列检测器就不需要这一步了。 4、还是得找一些资料,看看类似的样品的流动相,以它为起始流动相进行选择,如果没有,那就找专业书,看看它是怎么教你选择流动相的。 5、通过专属性验证所选色谱条件能否将各杂质与被测物分离检出 。应按0.5%(w/w)被测物浓度的各杂质量添加至被测物中,模拟被测物中可能存在杂质的状态,即有少量(约0.5%)杂质存在时能否与被测物达到完全分离(分离度大于2.0),以验证系统适用性。二、最低检测限 得出达到S/N=2或S/N=3时的样品药品浓度。三、供试品溶液浓度的确定 根据最低检出浓度,采用“上推法”来确定供试品溶液浓度:如一般设定杂质总量小于1.0%对照液,对照溶液的浓度至少应为最低检出浓度的20---50倍,供试品溶液浓度则应是最低检出浓度的2000~5000倍。同时还应考虑仪器、色谱柱等因素对最低检出浓度和最大进样浓度的影响(即耐用性因素),所以供试品溶液的浓度应在保证小于最大进样量的情况下,适当设定得高些,以保证该浓度在任何试验条件下,均有足够的检测灵敏度。四、线性及精密度试验 在稳定性考察中,如某杂质含量不断增加,则说明被测物降解的途径稳定、可循,则有必要对该杂质进行针对性地监控,即采用该杂质对照品 (经确证结构后,由人工合成获得)以外标法准确测定。此时,与含量测定相似,应进行线性试验。通常将杂质线性验证范围0.05%~1.5%)。且精密度试验也应符合要求,但RSD可根据实际情况,适当放宽至2.0~3.0。五、加样回收率试验 回收率试验采用在已知杂质含量的被测物中加入定量杂质的方法来评价。将各杂质以0.4%、0.5%、0.6% (w/w)的浓度加至被测物溶液中,以验证所采用的色谱条件是否可分离检测相应的各杂质以及与被测物中已存在的杂质是否累加,并观测累加量的准确性。六、强力破坏试验 强力破坏试验条件有强光、高温、高湿、强酸、强碱、氧化破坏等。破坏时不能过于剧烈,一般以产生20%~3O%杂质的条件为宜(《化学药物杂质研究技术指导原则》,国家食品药品监督管理局药品审评中心)。同时,还可采用二极管阵列检测器(DAD)或质谱(MS)监测器进一步验证主峰纯度,观察主峰中是否包含有被破坏产生的杂质峰。七、其他色谱参数的确定 流速的选择:主峰保留时间应在l0min以后,这样主峰不易出现拖尾、堆积的现象。柱温的选择:不应超过35℃,既可增强被测物与杂质的分离度,也可避免因温度过高使主峰降解,导致测定误差。溶剂的选择:由于有关物质测定的供试品溶液浓度高,应首先考虑被测物在溶剂中的稳定性 和溶解性。一般选流动相作溶剂为宜, 以排除溶剂峰的干扰。但由于有关物质的测定必须扣除溶剂峰,因此只要溶剂峰不干扰被测物质峰,即使不选用流动相作溶剂,也完全可以。在流动相溶解性不佳时,可采用甲醇或乙腈作溶剂以提高溶解性。 色谱图记录时间的设定:应能洗脱出有可能存在的全部杂质和经强力破坏试验产生的杂质,并规定至主成分保留时间的几倍为止。在测定制剂的有关物质时,个别辅料出峰滞后,此时应在质量标准中注明。八、杂质的限量。 必须根据《化学药物杂质研究技术指导原则》来定。
[b]高效液相色谱法的计算方法[/b]高效液相色谱法是用高压输液泵将具有不同极性的单一溶剂或不同比例的混合溶剂、缓冲液等流动相泵入装有固定相的色谱柱,经进样阀注入供试品,由流动相带入柱内,在柱内各成分被分离后,依次进入检测器,色谱信号由记录仪或积分仪记录。 1.对仪器的一般要求 所用的仪器为高效液相色谱仪。色谱柱的填料和流动相的组分应按各品种项下的规定.常用的色谱柱填料有硅胶和化学键合硅胶。后者以十八烷基硅烷键合硅胶最为常用,辛基键合硅胶次之,氰基或氨基键合硅胶也有使用;离子交换填料,用于离子交换色谱;凝胶或玻璃微球等,用于分子排阻色谱等。注样量一般为数微升。除另有规定外,柱温为室温,检测器为紫外吸收检测器。 在用紫外吸收检测器时,所用流动相应符合紫外分光光度法(附录Ⅳ A)项下对溶剂的要求。 正文中各品种项下规定的条件除固定相种类、流动相组分、检测器类型不得任意改变外,其余如色谱柱内径、长度、固定相牌号、载体粒度、流动相流速、混合流动相各组分的比例、柱温、进样量、检测器的灵敏度等,均可适当改变, 以适应具体品种并达到系统适用性试验的要求。一般色谱图约于20分钟内记录完毕。 2.系统适用性试验 按各品种项下要求对仪器进行适用性试验,即用规定的对照品对仪器进行试验和调整,应达到规定的要求;或规定分析状态下色谱柱的最小理论板数、分离度和拖尾因子. (1) 色谱柱的理论板数(n) 在选定的条件下,注入供试品溶液或各品种项下规定的内标物质溶液,记录色谱图,量出供试品主成分或内标物质峰的保留时间t(以分钟或长度计,下同,但应取相同单位)和半高峰宽(W),按n=5.54(t/W)计算色谱柱的理论板数,如果测得理论板数低于各品种项下规定的最小理论板数,应改变色谱柱的某些条件(如柱长、载体性能、色谱柱充填的优劣等),使理论板数达到要求。 (2) 分离度 定量分析时,为便于准确测量,要求定量峰与其他峰或内标峰之间有较好的分离度。分离度(R)的计算公式为: 2(t-t) R= ──W+W 式中 t为相邻两峰中后一峰的保留时间; t为相邻两峰中前一峰的保留时间; W及W为此相邻两峰的峰宽。 除另外有规定外,分离度应大于1.5。 (3) 拖尾因子 为保证测量精度,特别当采用峰高法测量时,应检查待测峰的拖尾因子(T)是否符合各品种项下的规定,或不同浓度进样的校正因子误差是否符合要求。拖尾因子计算公式为: W T=────── 2d 式中 W为0.05峰高处的峰宽; d为峰极大至峰前沿之间的距离。 除另有规定外,T应在0.95~1.05间。 也可按各品种校正因子测定项下,配制相当于80%、100%和120%的对照品溶液,加入规定量的内标溶液,配成三种不同浓度的溶液,分别注样3次,计算平均校正因子,其相对标准偏差应不大于2.0%。 3.测定法 定量测定时,可根据样品的具体情况采用峰面积法或峰高法。但用归一法或内标法测定杂质总量时,须采用峰面积法。 (1) 面积归一化法 测定供试品(或经衍生化处理的供试品)中各杂质及杂质的总量限度采用不加校正因子的峰面积归一法。计算各杂质峰面积及其总和,并求出占总峰面积的百分率。但溶剂峰不计算在内。色谱图的记录时间应根据各品种所含杂质的保留时间决定,除另有规定外,可为该品种项下主成分保留时间的倍数。 (2) 主成分自身对照法 当杂质峰面积与成分峰面积相差悬殊时,采用主成分自身对照法。在测定前,先按各品种项下规定的杂质限度,将供试品稀释成一定浓度的溶液作为对照溶液,进样,调节检测器的灵敏度或进样量,使对照溶液中的主成分色谱峰面积满足准确测量要求。然后取供试品溶液,进样,记录时间,除另有规定外,应为主成分保留时间的倍数。根据测得的供试品溶液的各杂质峰面积及其总和并和对照溶液主成分的峰面积比较,计算杂质限度。 (3) 内标法测定供试品中杂质的总量限度 采用不加校正因子的峰面积法。取供试品,按各品种项下规定的方法配制不含内标物质的供试品溶液,注入仪器,记录色谱图I 再配制含有内标物质的供试品溶液,在同样的条件下注样,记录色谱图Ⅱ。记录的时间除另有规定外,应为该品种项下规定的内标峰保留时间的倍数,色谱图上内标峰高应为记录仪满标度的30%以上,否则应调整注样量或检测器灵敏度。 如果色谱图Ⅰ中没有与色谱图Ⅱ上内标峰保留时间相同的杂质峰,则色谱图Ⅱ中各杂质峰面积之和应小于内标物质峰面积(溶剂峰不计在内)。如果色谱图Ⅰ中有与色谱图Ⅱ上内标物质峰保留时间相同的杂质峰,应将色谱图Ⅱ上的内标物质峰面积减去色谱图Ⅰ中此杂质峰面积,即为内标物质峰的校正面积;色谱图Ⅱ中各杂质峰总面积加色谱图Ⅰ中此杂峰面积,即为各杂质峰的校正总面积,各杂质峰的校正总面积应小于内标物质峰的校正面积。 (4) 内标法加校正因子测定供试品中某个杂质或主成分含量 按各品种项下的规定,精密称(量)取对照品和内标物质,分别配成溶液,精密量取各溶液,配成校正因子测定用的对照溶液,取一定量注入仪器,记录色谱图,测量对照品和内标物质的峰面积或峰高,按下式计算校正因子: A/m 校正因子(f)=─ A/m 式中 A为内标物质的峰面积或峰高;A为对照品的峰面积或峰高; m为加入内标物质的量; m为加入对照品的量。 再取各品种项下含有内标物质的供试品溶液,注入仪器,记录色谱图,测量供试品(或其杂质)峰和内标物质的峰面积或峰高,按下式计算含量:A 含量(m)=f×──A/m 式中 A为供试品(或其杂质)峰面积或峰高;m为供试品(或其杂质)的量; f、A和m的意义同上。 当配制校正因子测定用的对照溶液和含有内标物质的供试品溶液使用同一份内标物质溶液时,则配制内标物质溶液不必精密称(量)取。 (5) 外标法测定供试品中某个杂质或主成分含量,按各品种项下的规定,精密称(量)取对照品和供试品,配制成溶液,分别精密取一定量,注入仪器,记录色谱图,测量对照品和供试品待测成分的峰面积(或峰高),按下式计算含量:A 含量(m)=m×── A 式中各符号意义同上由于微量注射器不易精确控制进样量,当采用外标法测定供试品中某杂质或主成分含量时,以定量环进样为好。来源:分析化学网。[em61]
高效液相色谱法的计算方法高效液相色谱法是用高压输液泵将具有不同极性的单一溶剂或不同比例的混合溶剂、缓冲液等流动相泵入装有固定相的色谱柱,经进样阀注入供试品,由流动相带入柱内,在柱内各成分被分离后,依次进入检测器,色谱信号由记录仪或积分仪记录。1.对仪器的一般要求所用的仪器为高效液相色谱仪。色谱柱的填料和流动相的组分应按各品种项下的规定.常用的色谱柱填料有硅胶和化学键合硅胶。后者以十八烷基硅烷键合硅胶最为常用,辛基键合硅胶次之,氰基或氨基键合硅胶也有使用;离子交换填料,用于离子交换色谱;凝胶或玻璃微球等,用于分子排阻色谱等。注样量一般为数微升。除另有规定外,柱温为室温,检测器为紫外吸收检测器。在用紫外吸收检测器时,所用流动相应符合紫外分光光度法(附录Ⅳ A)项下对溶剂的要求。正文中各品种项下规定的条件除固定相种类、流动相组分、检测器类型不得任意改变外,其余如色谱柱内径、长度、固定相牌号、载体粒度、流动相流速、混合流动相各组分的比例、柱温、进样量、检测器的灵敏度等,均可适当改变,以适应具体品种并达到系统适用性试验的要求。一般色谱图约于20分钟内记录完毕。2.系统适用性试验按各品种项下要求对仪器进行适用性试验,即用规定的对照品对仪器进行试验和调整,应达到规定的要求;或规定分析状态下色谱柱的最小理论板数、分离度和拖尾因子.(1) 色谱柱的理论板数(n)在选定的条件下,注入供试品溶液或各品种项下规定的内标物质溶液,记录色谱图,量出供试品主成分或内标物质峰的保留时间t(以分钟或长度计,下同,但应取相同单位)和半高峰宽(W),按n=5.54(t/W)计算色谱柱的理论板数,如果测得理论板数低于各品种项下规定的最小理论板数,应改变色谱柱的某些条件(如柱长、载体性能、色谱柱充填的优劣等),使理论板数达到要求。(2) 分离度定量分析时,为便于准确测量,要求定量峰与其他峰或内标峰之间有较好的分离度。分离度(R)的计算公式为: 2(t-t) R= ──W+W 式中 t为相邻两峰中后一峰的保留时间; t为相邻两峰中前一峰的保留时间; W及W为此相邻两峰的峰宽。 除另外有规定外,分离度应大于1.5。(3) 拖尾因子 为保证测量精度,特别当采用峰高法测量时,应检查待测峰的拖尾因子(T)是否符合各品种项下的规定,或不同浓度进样的校正因子误差是否符合要求。拖尾因子计算公式为: W T=────── 2d 式中 W为0.05峰高处的峰宽; d为峰极大至峰前沿之间的距离。 除另有规定外,T应在0.95~1.05间。 也可按各品种校正因子测定项下,配制相当于80%、100%和120%的对照品溶液,加入规定量的内标溶液,配成三种不同浓度的溶液,分别注样3次,计算平均校正因子,其相对标准偏差应不大于2.0%。3.测定法 定量测定时,可根据样品的具体情况采用峰面积法或峰高法。但用归一法或内标法测定杂质总量时,须采用峰面积法。 (1) 面积归一化法 测定供试品(或经衍生化处理的供试品)中各杂质及杂质的总量限度采用不加校正因子的峰面积归一法。计算各杂质峰面积及其总和,并求出占总峰面积的百分率。但溶剂峰不计算在内。色谱图的记录时间应根据各品种所含杂质的保留时间决定,除另有规定外,可为该品种项下主成分保留时间的倍数。(2) 主成分自身对照法当杂质峰面积与成分峰面积相差悬殊时,采用主成分自身对照法。在测定前,先按各品种项下规定的杂质限度,将供试品稀释成一定浓度的溶液作为对照溶液,进样,调节检测器的灵敏度或进样量,使对照溶液中的主成分色谱峰面积满足准确测量要求。然后取供试品溶液,进样,记录时间,除另有规定外,应为主成分保留时间的倍数。根据测得的供试品溶液的各杂质峰面积及其总和并和对照溶液主成分的峰面积比较,计算杂质限度。(3) 内标法测定供试品中杂质的总量限度采用不加校正因子的峰面积法。取供试品,按各品种项下规定的方法配制不含内标物质的供试品溶液,注入仪器,记录色谱图I 再配制含有内标物质的供试品溶液,在同样的条件下注样,记录色谱图Ⅱ。记录的时间除另有规定外,应为该品种项下规定的内标峰保留时间的倍数,色谱图上内标峰高应为记录仪满标度的30%以上,否则应调整注样量或检测器灵敏度。如果色谱图Ⅰ中没有与色谱图Ⅱ上内标峰保留时间相同的杂质峰,则色谱图Ⅱ中各杂质峰面积之和应小于内标物质峰面积(溶剂峰不计在内)。如果色谱图Ⅰ中有与色谱图Ⅱ上内标物质峰保留时间相同的杂质峰,应将色谱图Ⅱ上的内标物质峰面积减去色谱图Ⅰ中此杂质峰面积,即为内标物质峰的校正面积;色谱图Ⅱ中各杂质峰总面积加色谱图Ⅰ中此杂峰面积,即为各杂质峰的校正总面积,各杂质峰的校正总面积应小于内标物质峰的校正面积。(4) 内标法加校正因子测定供试品中某个杂质或主成分含量按各品种项下的规定,精密称(量)取对照品和内标物质,分别配成溶液,精密量取各溶液,配成校正因子测定用的对照溶液,取一定量注入仪器,记录色谱图,测量对照品和内标物质的峰面积或峰高,按下式计算校正因子: A/m 校正因子(f)=─ A/m 式中 A为内标物质的峰面积或峰高;A为对照品的峰面积或峰高; m为加入内标物质的量; m为加入对照品的量。再取各品种项下含有内标物质的供试品溶液,注入仪器,记录色谱图,测量供试品(或其杂质)峰和内标物质的峰面积或峰高,按下式计算含量:A 含量(m)=f×──A/m 式中 A为供试品(或其杂质)峰面积或峰高;m为供试品(或其杂质)的量; f、A和m的意义同上。当配制校正因子测定用的对照溶液和含有内标物质的供试品溶液使用同一份内标物质溶液时,则配制内标物质溶液不必精密称(量)取。(5) 外标法测定供试品中某个杂质或主成分含量,按各品种项下的规定,精密称(量)取对照品和供试品,配制成溶液,分别精密取一定量,注入仪器,记录色谱图,测量对照品和供试品待测成分的峰面积(或峰高),按下式计算含量:A 含量(m)=m×── A 式中各符号意义同上由于微量注射器不易精确控制进样量,当采用外标法测定供试品中某杂质或主成分含量时,以定量环进样为好。来源:分析化学网。
[align=center][b]浅谈药典中高效液相色谱法测定中药材含量方法学验证的方法[/b][/align][align=center]西安国联质量检测技术股份有限公司[/align][align=center]食品事业部:任阳阳[/align]药典中关于方法学验证有相应的指导原则,指导原则上写的内容较宽泛和笼统,但对于大多数检测人员来说根本不知道如何下手,本文是根据自己的理解和经验来阐述如何做好药典中高效液相色谱的方法学验证。首先要了解做好药典中高效液相色谱的方法学验证,要好好的理解两部分的内容:第一,要熟悉0512高效液相色谱法这部分的内容;第二是理解9101药品质量标准分析方法验证指导原则,结合这两部分内容,做好方法学验证。1. 确定本实验室要使用的仪器和配件能否满足本次验证的色谱提交,包括色谱柱、检测器和流动相。2. 0512方法中验证系统适用性实验首先要了解色谱的适用性实验包括5个参数:理论塔板数、分离度、灵敏度、拖尾因子和重复性。2.1理论塔板数和分离度在已调试好的色谱条件下,分析目标物质,根据分析结果计算此方法的理论塔板数和分离度。一般待测物质色谱峰与相邻色谱峰之间的分离度不大于1.5。2.2 灵敏度通过一系列浓度的对照品确定信噪比,其灵敏度要求定量测定时信噪比≥10;若为定性鉴别其信噪比要求≥3。2.3拖尾因子T拖尾因子是评价色谱峰的对称性结果,以T表示,对对照品进行进样,分析色谱结果,以峰高作定量参数时,T值应在0.95~1.05之间。2.4 重复性 一般采用外标法,取对照品溶液连续进样5次,计算5次进样的峰面积的相关标准偏差,要求≤2.0%以上5点是0512方法内容中需要验证的5点内容,已对方法进行了简单的验证,但这些不够,还得结果9101的内容进行详细的进一步的验证。3. 9101验证的指标主要有准确度、精密度、专属性、检出限、定量限、线性和耐用性,其中精密度可以参考0512中已验证的重复性的检测结果;检出限和定量限也可参考0512中验证的结果。3.1准确度准确度是指该方法测定结果与真实值或参考值的接近程度,常用回收率进行验证。一般选择高、中、低三个浓度进行测定,也就是说,对照品加入量与待测供试品含量的比值为1.5:1,1:1和0.5:1进行加标实验,分析加标结果,计算回收率和回收率的相对标准偏差,结果必须满足药典要求。3.2 专属性向试样中加入杂质或辅料,与杂质和辅料的结果进行比较,确定测定结果是否受干扰。3.3 线性根据待测成分的含量大小,选择至少5份合适的浓度范围进行分析,以测得的响应信号(峰高或峰面积)对被测物的浓度作图,求出线线方程和相关系数。3.4耐用性一般经过变动流动相的比例、流动相的pH、不同类型的色谱柱。柱温以及流速来进行验证,验证测定条件发生微小变化时,测定结果不受影响的程度,从而确保方法的可靠性。综上所述,以上是高效液相色谱法测定中药材含量的方法学验证的所有指标,通过以上指标的完善和验证,才能做出一套完成的方法学验证。
[align=center][size=18px][b]二维液相色谱法的杂质分析[/b][/size][/align][align=left]一、 目的:[/align][align=left]利用二维液相色谱法对氟氯西林钠的聚合物杂质进行定性分析,确定凝胶色谱分离的第一个杂质峰与有关物质分析方法(反向色谱分析)中的得到第一个杂质峰为同一个物质。[/align][align=left]二、实验仪器:[/align][align=left]电子天平 型号:T-214 [/align][align=left]pH计 型号: PB-10[/align][align=left]液相色谱仪 型号:U3000 (双三元 高压泵 柱温箱附带十通切换阀)[/align][align=left]100 μl定量环[/align][align=left]三、 实验用试剂:[/align][align=left]磷酸二氢钠、磷酸氢二钠、、磷酸二氢钾、磷酸、甲醇、乙腈、实验用水(mili-Q)[/align][align=left]四、 实验样品:[/align][align=left]氟氯西林钠(制剂提供)[/align][align=left]五、 色谱条件:[/align][align=left]1、 右泵 (凝胶色谱)[/align][align=left]色谱柱:Tsk G2000SW XL(7.8mm*300mm 5μm)[/align][align=left]流动相:0.04mol/l磷酸二氢钠-0.04mol/l磷酸氢二钠(61:39)pH=5.0:乙腈(95:5)[/align][align=left]VWD检测器波长:215nm 进样量:20μl 流速:0.8ml/min 柱温:25[font='宋体']℃[/font][/align][align=left]2、 [font='宋体']左泵 (反向色谱)[/font][/align][align=left][font='宋体']色谱柱:Agela Venusil MP C[/font][font='宋体'][size=13px]18[/size][/font][font='宋体'] [/font](4.6mm*250mm 5μm)[/align][align=left]流动相A:乙腈 -2.7g/磷酸二氢钾溶液(pH=5.0)=20:80[/align][align=left]流动相B:乙腈 -2.7g/磷酸二氢钾溶液(pH=5.0)=40:60[/align][align=left]按下表程序梯度洗脱:[/align][table][tr][td][align=center][font='calibri'][size=13px]时间([/size][/font][font='calibri'][size=13px]min[/size][/font][font='calibri'][size=13px])[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]流动相[/size][/font][font='calibri'][size=13px]A[/size][/font][font='calibri'][size=13px]([/size][/font][font='calibri'][size=13px]%[/size][/font][font='calibri'][size=13px])[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]流动相[/size][/font][font='calibri'][size=13px]B[/size][/font][font='calibri'][size=13px]([/size][/font][font='calibri'][size=13px]%[/size][/font][font='calibri'][size=13px])[/size][/font][/align][/td][/tr][tr][td][align=center][font='calibri'][size=13px]0[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]100[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]0[/size][/font][/align][/td][/tr][tr][td][align=center][font='calibri'][size=13px]10[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]100[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]0[/size][/font][/align][/td][/tr][tr][td][align=center][font='calibri'][size=13px]30[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]50[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]50[/size][/font][/align][/td][/tr][tr][td][align=center][font='calibri'][size=13px]40[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]0[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]100[/size][/font][/align][/td][/tr][tr][td][align=center][font='calibri'][size=13px]50[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]0[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]100[/size][/font][/align][/td][/tr][tr][td][align=center][font='calibri'][size=13px]55[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]100[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]0[/size][/font][/align][/td][/tr][tr][td][align=center][font='calibri'][size=13px]65[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]100[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]0[/size][/font][/align][/td][/tr][/table][align=left][/align][align=left]六、 实验过程:[/align][align=left]1、 首相使用右泵接凝胶色谱柱测得聚合物色谱图,由此色谱图计算确定二维反相色谱的阀切换时间。[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008261816118191_9119_2204446_3.png[/img][/align][align=left]由图可知:第一个杂质峰的保留时间为16.517min 流速0.8ml 截取100μl 所用的时间就等于1/8=0.125min,所以初步确定阀切换时间为:16.517-0.125=16.392min.[/align][align=left]2、 使用左泵的反相系统色谱条件测得杂质的保留时间为:4.301min见下图[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008261816122917_6408_2204446_3.png[/img][/align][align=left][/align][align=left]3、 编辑采集方法1设置阀切换时间为16.392min,编辑程序关键点如下图:[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008261816124236_7198_2204446_3.png[/img][/align][align=left]起始阀的位置是10[font='calibri'][size=13px] [font=calibri]—[/font][/size][/font]1,在第二行追加命令为16.392min阀的位置切换至1 [font='calibri'][size=13px]—[/size][/font]2。左泵、右泵同时运行,当右运行至16.392min时阀切换至1 [font=calibri][size=13px]—[/size][/font] 2,截取通过凝胶色谱分析的聚合物100μl 至反相色谱系统中,依左泵程序梯度洗脱。得到相对保留时间一致的杂质峰。[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008261816125281_6542_2204446_3.png[/img] [/align][align=left]10[font=calibri][size=13px] ---[/size][/font]1右泵连接示意图[/align][align=left] [img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008261816128650_7051_2204446_3.png[/img][/align][align=left] 1[font='calibri'][size=13px]——[/size][/font]2左泵管路示意图[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008261816129812_7633_2204446_3.png[/img] [/align][align=left]利用药瓶盖二维并联连接管路示意图 [/align][align=left] [img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008261816130652_3475_2204446_3.png[/img][/align][align=left] 二维色谱图 [/align][align=left][b]七、结论[/b]:二维液相色谱杂质色谱图表留时间为21.796min-16.392=5.0404min,相对保留时间在可接受范围内。[/align][align=left][b]注意事项[/b]:[/align][align=left]1、熟悉仪器管路基本连接,明确实验目的。[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008261816131687_577_2204446_3.png[/img][/align][align=left]改装后的并接流路图,白色的为新增加的定量环。[/align][align=left]3、 阀切换的时间确定一定要以实验数据为依据,可截取聚合物保留时间的前后两端时间,来确定最终的阀切换时间。[/align][align=left]修改后的采集方法左泵反相梯度洗脱应该适当调整,以本次实验为例最终采集时间延长16min,编辑采集方法时要确定[/align][align=left]阀切换的位置和时间。[/align][align=left]4、 右泵凝胶色谱流路,采集时间延长至81min。[/align][align=left]5、 调整后的左泵梯度洗脱程序如下:[/align][table][tr][td][align=center][font='calibri'][size=13px]时间([/size][/font][font='calibri'][size=13px]min[/size][/font][font='calibri'][size=13px])[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]流动相[/size][/font][font='calibri'][size=13px]A[/size][/font][font='calibri'][size=13px]([/size][/font][font='calibri'][size=13px]%[/size][/font][font='calibri'][size=13px])[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]流动相[/size][/font][font='calibri'][size=13px]B[/size][/font][font='calibri'][size=13px]([/size][/font][font='calibri'][size=13px]%[/size][/font][font='calibri'][size=13px])[/size][/font][/align][/td][/tr][tr][td][align=center][font='calibri'][size=13px]16[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]100[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]0[/size][/font][/align][/td][/tr][tr][td][align=center][font='calibri'][size=13px]26[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]100[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]0[/size][/font][/align][/td][/tr][tr][td][align=center][font='calibri'][size=13px]40[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]50[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]50[/size][/font][/align][/td][/tr][tr][td][align=center][font='calibri'][size=13px]56[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]0[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]100[/size][/font][/align][/td][/tr][tr][td][align=center][font='calibri'][size=13px]66[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]0[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]100[/size][/font][/align][/td][/tr][tr][td][align=center][font='calibri'][size=13px]71[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]100[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]0[/size][/font][/align][/td][/tr][tr][td][align=center][font='calibri'][size=13px]81[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]100[/size][/font][/align][/td][td][align=center][font='calibri'][size=13px]0[/size][/font][/align][/td][/tr][/table]
有没有分析3-氨基丙腈的液相色谱分析方法我想用液相色谱分析有机盐中的3-氨基丙腈的含量,但由于3-氨基丙腈在紫外和示差液相中响应值不是很大,满足不了微量杂质的分析。大家有没有好的建议。比如有没有合适的检测器、或则有合适的衍生法。(由于样品中含有氨基丙酸,所以氨基的衍生手段好像无法使用)
最近我按照《GBT 23884-2009 动物源性饲料中生物胺的测定 高效液相色谱法》测定鱼粉饲料中的组胺,结果一直不理想,标准品衍生化反应还好,峰形较好,且线性关系良好,但是加标质控样品一衍生上机后,就出现有的样品目标峰前面老是有个貌似是杂峰一直和目标峰分不开,而有的平行样品却没有这个杂峰出现,加标回收也不高。 请问群友,这个是什么原因呀?大家又是用什么方法做的呢?谢谢指教!

我在本网站看到【小叶子集结号】的色谱柱试用活动,就抱着试试的心理报了名。过了段时间接到活动举办方的信息确认电话,信息确认三天后就收到对方通过顺丰快递邮寄的色谱柱(速度真快),规格是SCINChrom C18G 5μm 150mm*4.6mm。http://ng1.17img.cn/bbsfiles/images/2017/01/201701191700_668190_2514846_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016060316154455_01_2514846_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016060316155811_01_2514846_3.jpg我公司主要产品是一种除草剂原药,其中有项检测指标是低极性杂质,指标要求<0.1%,最初进行分析方法确认时更换多个厂家色谱柱均不能满足检测要求,最终经过多方咨询才确认杂质检测色谱柱。试用SCINChrom C18G 5μm 150mm*4.6mm 色谱柱 完全能满足我公司产品检测要求。分析条件:乙腈:甲醇:水=27:39:34,保留时间60min。谱图见下图SCINChrom C18G 5μm 150mm*4.6mmhttp://ng1.17img.cn/bbsfiles/images/2016/06/201606031647_595936_2514846_3.jpg公司目前使用色谱柱谱图(报废色谱柱)http://ng1.17img.cn/bbsfiles/images/2016/06/201606031647_595938_2514846_3.jpg检测仪器为岛津液相色谱仪http://ng1.17img.cn/bbsfiles/images/2016/06/201606031647_595939_2514846_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/06/201606031647_595940_2514846_3.jpgSCINChrom C18G 5μm 150mm*4.6mm色谱柱 完全能满足我公司产品的严格检测条件,是一款非常不错的产品,值得推荐。最后谢谢仪器信息网举办的试用活动。
多酚类物质使用质谱法测定的较多,这主要是避免了杂质干扰,但从仪器配备的广泛性和准确定量的角度,[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法是比较容易接受的方法。文献报道了利用Hypersil Gold柱,流动相为1%乙酸和甲醇,梯度洗脱,1针70min,流速0.8 ml/min,进样量5微升,建立了没食子酸、新绿原酸、(+)-儿茶素水合物、绿原酸、香草酸、咖啡酸、丁香酸、(-)-表儿茶素、对香豆酸、阿魏酸、芥子酸酸、水杨酸、鞣花酸、芦丁、杨梅素、槲皮素和山奈酚16种多酚类物质的[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]方法。在仪器配备和方法上可操作性强,能为相关研究者提供有益借鉴。详见[url]https://doi.org/10.3390/horticulturae8020084[/url]
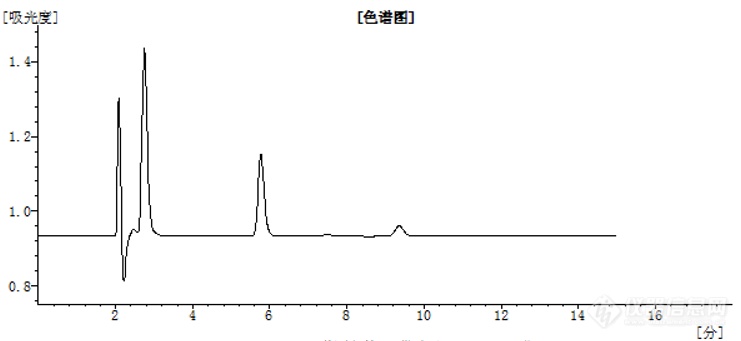
【求助】阿司匹林液相色谱图在两分钟左右有多个杂质峰还有一个很大的倒峰是怎么回事啊?还有液相色谱图长这样正常嘛?[img=,690,317]https://ng1.17img.cn/bbsfiles/images/2021/05/202105280909595087_3037_5279865_3.png!w690x317.jpg[/img][img=,549,281]https://ng1.17img.cn/bbsfiles/images/2021/05/202105280910096243_2274_5279865_3.png!w549x281.jpg[/img][img=,541,272]https://ng1.17img.cn/bbsfiles/images/2021/05/202105280910149427_6296_5279865_3.png!w541x272.jpg[/img][img=,690,310]https://ng1.17img.cn/bbsfiles/images/2021/05/202105280910207317_3122_5279865_3.png!w690x310.jpg[/img]
不溶性糖精的高效液相色谱测定方法?请问用高效液相色谱法测定不溶性糖精的具体测量条件,如流动相、流速等,越详细越好,谢谢!!!!
[color=#444444]我的一个实验原材料因为是聚合物,购买的原材料浓度应该在95%以上。现在我需要对实验对象中这种原材料的含量进行测定,但打了色谱后,发现原材料那里有杂质峰,推测这个杂质很可能就是聚合物的单体,是不是存在这个杂质峰我就无法做定量分析?请问有什么好办法可以解决这个问题吗?谢谢![/color]
[b][font=宋体]问题描述:如何采用液相色谱测定氨基酸?对仪器配置有哪些要求?有哪些注意事项?[/font][font=宋体]解答:[/font][/b][font=宋体]([/font]1[font=宋体])液相色谱通常无法直接测定氨基酸,需要对氨基酸进行衍生,衍生的方式可采用柱前衍生和柱后衍生,柱前衍生有专门的试剂包,许多分析仪器厂家有相关的应用资料;柱后衍生需要相应的衍生装置和衍生试剂。[/font][font=宋体]([/font]2[font=宋体])仪器配置方面,液相色谱仪首先需要具有梯度洗脱功能;防止温度波动导致保留时间的漂移,因此需要配备柱温箱。氨基酸衍生产物具有紫外和荧光响应,可以配置紫外检测器、二极管阵列检测器或荧光检测器进行检测。如果采用在线柱后衍生方式进行测定,还需配备柱后衍生装置。[/font][font=宋体]([/font]3[font=宋体])由于氨基酸测定种类较多(通常是[/font]18[font=宋体]种游离氨基酸),另外样品基质较为复杂,采用液相色谱测定氨基酸时,通常会出现保留时间漂移、分离度下降、杂质干扰等现象。[/font][font=宋体]([/font]4[font=宋体])液相色谱法测定食品中的氨基酸含量,可以参考:[/font]SN/T 5223-2019[font=宋体]《蜂蜜中[/font]18[font=宋体]种游离氨基酸的测定[/font][font=宋体]高效液相色谱[/font][font=宋体]-[/font][font=宋体]荧光检测法》、[/font]QB/T4356-2012[font=宋体]《黄酒中游离氨基酸的测定[/font][font=宋体]高效液相色谱法》。[/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font]
蜂王浆样品用水溶解后经正已烷-二氯甲烷(1:1,V/V)提取,无水乙醇破除乳化,样液经离心后,用反相固相萃取柱富集与净化,用乙腈洗脱后,氮吹至近干,用80%乙腈水溶液溶解,经0.22μm滤膜过滤后,采用超高效液相色谱梯度洗脱与紫外检测器分析蜂王浆中的氟胺氰菊酯与氟氯苯氰菊酯残留。以XTerra Shield RP18柱(2.1mm×50mm,1.7μm)为色谱柱,以水和乙腈为流动相,两种菊酯残留在4min中内实现了较好的分离,方法快速准确,灵敏度高,是一种较好的定性和定量方法。所研究的提取净化方法及色谱分离条件能有效排除蜂王浆中的杂质干扰,添加回收率在75.7%~82.8%之间,方法检出限为10μg/kg。
[font=&][color=#666666] 对于初学者来说,分析液相色谱仪方法可能会让人不知所措。此类方法通常包含与实验中每个步骤相关的信息和说明。熟悉实验室中液相色谱仪方法的格式很重要,这样您就可以轻松找到整个过程的方法。[/color][/font][font=&][color=#666666]对液相色谱仪器进行设置[/color][/font][font=&][color=#666666] 准备好一台[url=https://www.risun-tec.com]液相色谱仪器[/url]、色谱柱和化学品,下一步就是开始设置您的仪器进行测试。检查液相色谱仪器的校准状态非常重要。所选仪器必须成功通过所有校准测试;这通常由仪器上的贴纸或标签指示,说明上次和下一次校准的结果和日期。进行校准测试以确保仪器的每个部分都在适当的范围内工作。例如,必须检查泵的流量以匹配数字输入;对检测器、进样器和色谱柱加热器进行了类似的测试。目的是确保实验室中的所有仪器都能正常工作并且在预定的误差范围内。[/color][/font][font=&][color=#666666] 选择仪器后你必须对其进行清洁并准备色谱柱。清洁步骤对于去除之前工作中的任何残留物很重要;因此可以获得稳定的基线和准确的结果。许多方法规定了在测试之前清洗系统的特殊顺序。通常,清洁程序包括先用流水去除任何缓冲液残留物,然后再用纯甲醇或异丙醇/甲醇混合物。最后,将 50% 的有机溶液冲洗通过系统。最终冲洗液的组成很大程度上取决于所使用的色谱类型:反相或正相。还根据色谱柱的类型和要使用的流动相的组成对色谱柱进行冲洗。查阅色谱柱的宣传单以确定适合您的色谱柱的最佳冲洗和储存解决方案。[/color][/font][font=&][color=#666666]用于液相色谱仪测试的溶液的制备[/color][/font][font=&][color=#666666] 首先要制备的溶液通常是流动相。有时,当使用梯度系统时,您需要为您的分析方法制备多个流动相。使用液相色谱仪级水和溶剂来制备流动相非常关键。准备好流动相后,就可以开始用相应的溶液冲洗系统并开始平衡色谱柱了。大多数 HPLC 系统本质上是四元系统,即可以从四个不同的入口管线泵送。如果要使用多条管线(即该方法涉及梯度),那么通过每条管线冲洗的最终溶液应该是与流动相兼容的溶剂。[/color][/font][font=&][color=#666666] 例如,如果流动相的缓冲液含量高,则入口管线必须用至少 50% 的冲洗水溶液冲洗,以防止流动相中的盐析出并堵塞入口管线。所有冲洗液和流动相都应通过微过滤器(0.45 μm 或 0.2 μm)过滤以去除细菌和有机碎片。[/color][/font][font=&][color=#666666] 冲洗系统并开始平衡色谱柱后(在某些情况下可能需要 3 小时或更长时间,具体取决于色谱柱类型和流动相组成),您应该开始处理样品和参考溶液。在许多情况下,您在制备这些溶液时需要采取特殊的预防措施,例如减少暴露在光线或湿气中的时间,以防止分析物降解并产生错误结果。这些特殊的注意事项通常会在分析方法中提及。参考溶液和样品溶液也应通过称为注射器过滤器的特殊过滤器进行过滤(因为它们安装在注射器上)。但是,所用过滤器的类型和材料应与分析物和溶剂兼容,以避免吸附或污染。[/color][/font][font=&][color=#666666]处理色谱图并计算出结果[/color][/font][font=&][color=#666666] 一旦您确定您的系统工作正常,请恢复进样顺序以进样样品溶液。完成序列后,您可以使用特定软件对采集的色谱图进行处理,从而得出有意义的结果。一些软件程序允许您“整合”所有峰,无论它们有多小。计算每个峰下的面积,并使用生成的数字来确定测试结果。液相色谱仪实验后有两种主要的计算方式:首先,通过使用测定来确定一种或多种活性成分的含量,例如计算片剂中扑热息痛的含量。或者,您可能希望确定色谱图,即确定杂质的类型及其相对比例。在您输入分子量和稀释因子后,当前的软件可以进行计算并生成最终报告。[/color][/font]







 我要推广仪器
我要推广仪器
 下载APP
下载APP