[color=#444444]合成的反应物送[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]检测,液相在1.8和2.2出两个峰,质谱检测分子量只差1,有可能是什么原因[/color]
[color=#444444]乙酰丙酸用液相色谱进行分析时,如果用268nm检测只有一个峰,在215nm下检测会出现两个峰,且两个峰时间间隔有近2min,有哪位大神能帮解释一下这是为什么啊[/color]
利用纯品的母离子和子离子构建了定量检测目标化合物的方法,标准品检测没问题,可检测样品时出现两个峰,两峰之间相差1分钟,请问这是问什么?
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法检测敌百虫,进了一针标准容易,前后出现两个峰。前面的小峰怀疑是敌敌畏,无论如何改变色谱条件都出现两个峰。这种情况下如何定量呢?目前色谱条件:进样口100;检测器180;柱温45,10℃/min,升至150,保持5min。
TM-5 ( 5%二苯基95% 二甲基聚硅氧烷),规格:30*0.25*0.25检测条件:60度保持1分钟,程序升温10度/分钟,至200度。发现99%纯度的环氧苯乙烷 一直出现两个峰,峰面积查不多,不像是杂质峰,两个峰的时间为4.9分钟和5.4分钟,开始以为是试剂的问题,后来换了5个厂家的环氧苯乙烷试剂,都出现同样情况。按道理,纯物质应该只有一个检测峰的吧!但是为何检测99%纯度的环氧苯乙烷时,会出现两个峰?
如题,液相分析含P化合物,紫外检测器,是否所有含P的化合物都会出两个峰呢?求助各位老师
我的化合物分子量是225,从核磁上看应该是我的化合物,纯度也在95以上,但做LCMS时,主峰的分子量却是243(正模式)。请问MS可能会出现M+水的峰吗?
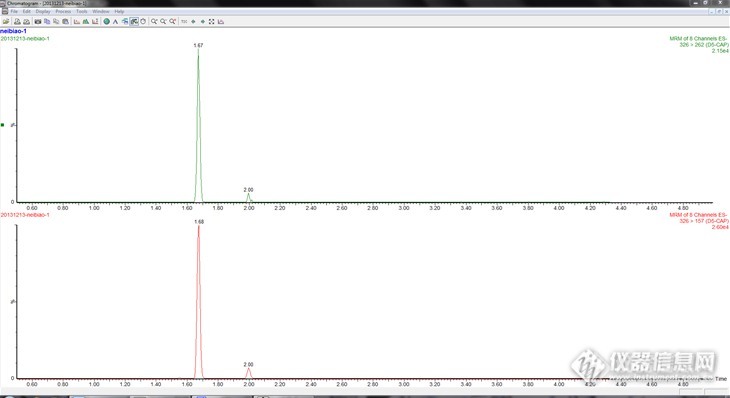
做液质联用是,用氘代氯霉素做内标,出现两个峰。就是在子离子通道也是两个峰。本来应该只有1.67出一个氘代氯霉素的峰啊,2.00那里出的是什么峰啊。而且262和157两个子离子通道都有这种情况。http://ng1.17img.cn/bbsfiles/images/2013/12/201312141813_482020_2698225_3.png是因为cl的相对分子量有35和37两个吗?
液相色谱负峰很正常,气相色谱怎么就不正常了?气相色谱哪些检测器是不会出现负峰的?为什么?出现负峰一般都是什么原因呢
各位: 今天跟同事讨论一问题,低分子量PEG 像PEG400 与高分子量固体状PEG,像PEG20000混溶会出现分层现象吗? 它们在室温下能互溶吗?
做某一些天然毒素时候,出现了同分异构体的峰,单标的时候也会出现两个质谱峰,求解!
在进样,检测过程中,忘记拧紧purge阀,仪器,数据等会出现什么问题及解决方法
如题,同碳上的两个氢会在H-H COSY里边出现交叉峰吗?另外,据说在HMBC里会出现“自旋峰”(不确定是不是这个名字哈),请问有这种情况吗?如果有,怎么鉴别啊?我新手,求解答!谢谢!!!
[color=#444444]如下图所示。所测化合物分子量只有122,为什么谱图里会出现接近300的质荷比???这张谱图该如何分析?流动相应该是甲醇,仪器是LCQ-FLEET。其余的就不太知道了。大神求助[/color][color=#444444][img]http://muchongimg.xmcimg.com/oss2/img/2018/0505/bw173h3877134_1525503506_542.png#opennewwindow[/img][/color]

我用液质联用检测抗生素,用中检所的标准品,单标居然出现两个峰,大家帮分析下是啥问题。目标物是阿莫西林,溶剂是甲醇/水(50/50),液相流动相是乙腈/水,梯度洗脱,20%-80%,时间18min。用头孢氨苄和头孢拉定的单标进样也都出现两个峰,纯度写的是95%。两张图分别为阿莫西林单标的质谱离子流图和两个峰处的碎皮离子峰图,两个峰的碎皮离子峰图是一样的。http://ng1.17img.cn/bbsfiles/images/2012/04/201204110831_360531_2424544_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/04/201204110832_360532_2424544_3.jpg
各位大侠:本人最近用waters液相色谱仪2174荧光检测器测一个衍生化样品时,不时有很大一个或两个倒峰出现,请问是些什么原因会出现这种现象呢?怎么注意或解决这一问题?急求赐教!谢谢!
TCD检测器,so1 pl1温度为130度,柱温150度,用标气测氢气时,在1.2分钟的时候连续出现两个峰不知道怎么回事,求大神解答
[color=#444444]做的化合物的高分辨率质谱,负离子模式的,分子量为949,怎么会出现这么多+46和+113,求高人解答一二[/color][color=#444444][img]http://muchongimg.xmcimg.com/oss2/img/2019/0218/bw156h8256704_1550469901_552.png[/img][/color]
是第一次做液质,得到了结果,可是有些地方不知道怎样去分析和考虑,比如有些对应单个总离子流色谱峰的分子量和对应液相色谱峰的分子量不一致,请教各位前辈,为什么会出现这种情况,怎么样去解决?我检测的过程中出现两个峰对应同一分子量的情况,无法确定化合物名称,我做的的类黄酮,可能是因为其中的羟基取代位置不同造成的,请问除了对应标准品的保留时间外,用什么仪器或者方法可以区分两者?先谢谢各位!
[color=#444444]我合成的杂环化合物,其中一个原料用到过氢氧化钠,在合成目标化合物的时候,发现其中部分化合物的基峰分子量竟然是M×2+23,也有M+1峰和M+23峰。[/color][color=#444444]合成化合物的结构含有较多的氮元素,尝试打氢谱,发现氢谱是对的[/color][color=#444444]。[/color][color=#444444]但是质谱出现M×2+23,却难以解释,专业人士多多指教![/color]
我有一个质谱图,化合物精确分子量为522.0266.。理论上的M+1 峰应该是 522.0300+1.0078=523.0378。现在正离子模式 质谱图上出现了522.0312(丰度100)和523.0270(丰度35)问题:图谱上523.0270应该是M+1峰,看丰度不应该是同位素造成的而是仪器造成的,问522.0312的峰如何解释,同一个质谱会同时出现M和M+1吗?
请问专家,我在用[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]测无机阴离子时,依据什么来确定离子的种类,是根据出峰的时间吗?为什么会出现两个峰连在一起而没有分开,这时又如何判断其种类,某一离子出峰的时间范围是多少呢?
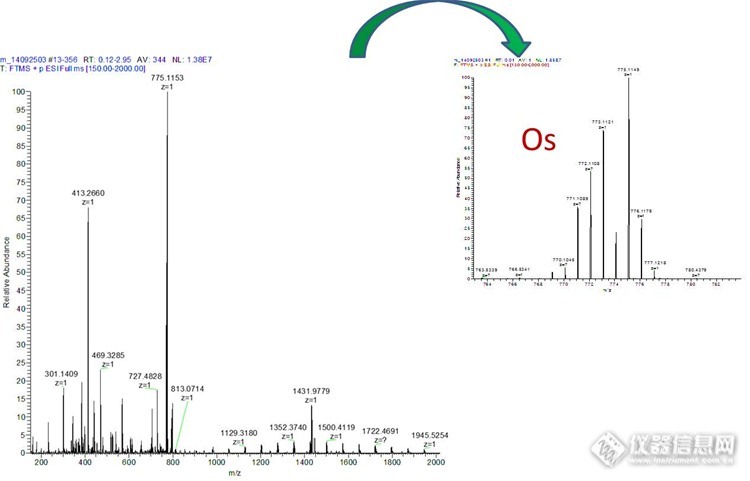
质谱应用之分子量测量 最近10年质谱技术的飞速发展,耐用的离子源,高性能的质量分析器和多种有效的扫描方式推动了质谱仪器走进各个单位,质谱成为功能强大的生物化学分析平台。目前基于质谱的物质定量定性实验应用广泛,从普通色谱-质谱(GC-LC&LC-MS)连用技术的定量分析实验(药理药代、农残筛查、环境污染物分析……),到大规模发现鉴定的组学实验(蛋白质组学和代谢组学)。抛开这些酷炫的方法和技术,我们今天讨论一下质谱的基本应用——测定分子量,通过一些测定分子量的实验我们可以看到分子量代表的更多意义。 质谱分析是一种测量离子质荷比(质量-电荷比)的分析方法,质谱法(Mass Spectrometry, MS)即用电场和磁场将运动的离子(带电荷的原子、分子或分子碎片,有分子离子、同位素离子、碎片离子、重排离子、多电荷离子、亚稳离子、负离子和离子-分子相互作用产生的离子)按它们的质荷比分离后进行检测的方法。测出离子准确质量即可确定离子的化合物组成。这是由于核素的准确质量是一多位小数,决不会有两个核素的质量是一样的,而且决不会有一种核素的质量恰好是另一核素质量的整数倍。分析这些离子可获得化合物的分子量、化学结构、裂解规律和由单分子分解形成的某些离子间存在的某种相互关系等信息(以上内容来自百度百科和高中教科书)。从定义我们看出,测定分子量是质谱的基本技能,一台质谱仪我们首先问的是它测量的分子量范围是多少,测量的准确度怎么样。1小分子的测定 质谱的首先发展是测定元素的相对分子量,比如我们一般说到元素C的分子量是12,其实说的是C在自然界的最高丰度12C的相对原子量,考虑自然界只有12C相对含量1.082%的13C,C准确平均分子量是12.011。化合物一般有C、H、O……多种元素组成,这些元素的同位素互相组合,如果我们的质谱可以区分相邻的同位素的相对分子量,质谱图上会显示的一簇峰,每个质谱峰对应相同的分子式下不同的同位素组成的化合物响应。因为化合物组形成元素的不同,他们的质谱簇峰分子量(momoisotopic mass)组成独特的质谱峰模式(pattern),如果质谱区分不了相邻的同位素峰,这一簇峰变成一个质谱峰所对应的是平均分子量(average mass)。 如果我们测定一个化合物分子量,如果通过质谱可以得到精细的元素分子量(momoisotopicmass)及其相对丰强度(在质谱上表现为簇峰的强度)的信息,可以通过谱图推测化合物的组成写出分子式。图1 A是测的城市污水提取物的分子量,三个主要质谱峰为同一个化合物的同位素质谱峰,推测分子式为C2HO2Br2,采用软件(很多软件都可以进行,最简单的是chem office)模拟此分子式的精确分子量,图2 B即为模拟所得的质谱图。可以看出所测得的质量偏差很小,最高元素峰216.8331-216.8328=0.0003Da,质谱峰分布模式(分子量和相对强度)实际测量图和模拟图几乎一致,可以确定该化合物的分子式是C2HO2Br2。http://ng1.17img.cn/bbsfiles/images/2014/12/201412131121_526995_2265735_3.jpg图1 污水提取物质谱图。A测量图,B模拟图。质谱Thermo LTQ-orbit,HESI源。 对于有特殊的元素的化合物,测量准确的分子量及其同位素质谱模式可以准确的判定特殊元素的存在,图2是测得某配位化合物的质谱图,通过其特殊的质谱图可以确定此化合物为Os金属配合物。http://ng1.17img.cn/bbsfiles/images/2014/12/201412131125_526996_2265735_3.jpg图2 Os配合物质谱图。质谱Thermo LTQ-orbit,HESI源。 上述测量过程简单实用,但是这个实验要求质谱有足够的质量准确度,所测的分子量与实际值最好在小数点最后一位有波动,不然预测分子式会有很大的偏差。2更高分子量的测量 对于同位素峰的测量,需要质谱区分相邻的同位素峰。在图1中两个同位素峰相差越2个道尔顿,在测量217分子量时候,只要质谱可以区分2个道尔顿的质谱峰就可以了,在图2中,同位素峰相差1道尔顿,区分度只有1个道尔顿。当分子量达到5K以上的时候,如果化合物仅仅由CHON等简单同位素组成,因为组成原子个数的增多,同位素峰越来越复杂,两个同位素峰之间的区分度越来越小,当质谱区分不开这些同位素峰的时候,测得是平均分子量(average mass)。图3 A测量的是一个分子量为10380Da的多肽,B和C是带10个电荷和11电荷同位素峰的局部方法图。在B中,同位素质谱峰间距(区分度)为0.1001Da。随着分子量的增加,需要质谱对相近同位素峰区分能力更强。评价质谱这种能力的指标是分辨率,我们一般用单位分辨率R=m/Δm来表示(该论述与严格定义有区别),图1需要的分辨率217/2=108,图2的分辨率780/1=780,而图三需要的分辨率1100/0.1=11000。所以说准确测分子量尤其是大分子量需要质谱具有高的分辨率。http://ng1.17img.cn/bbsfiles/images/2014/12/201412051959_526030_2265735_3.jpg图3多肽质谱测定。 A,质谱图B,,+10电荷质谱放大图C,+11电荷质谱放大图。Thermo LTQ-orbit,HESI源。3不同离子源的测定大分子的策略 目前测定大分子的主要离子源有基质辅助激光解吸(MALDI)和电喷雾(ESI)。图4是采用不同离子源测定聚乙二醇修饰药物分子量,A是MALDI质谱测得,几乎为所有分子的都带一个电荷,质谱间距为聚乙二醇重复单元-CH2-CH2-O-44Da;B为ESI质谱所测谱图,Z为分子所带电荷数,z=4质谱间距为44/4=11,z=3质谱间距为44/3=14.67。http://ng1.17img.cn/bbsfiles/images/2014/12/201412052001_526031_2265735_3.jpg图4聚乙二醇化药物质谱图。A AB MALDI-TOF谱图,基质DHB反射模式;B Thermo ESI-LTQ-Orbit谱图。 MALDI电离的离子一般带一个电荷(随着分子量增加,会出现带多个电荷的情况),图5是测得8478和11675多肽质谱图,5737为11675多肽带双电荷所得。采用MALDI测量分子量谱图测量结果直观方便,图6是测量分子
[color=#444444]液相色谱质谱联用时,比如在液相色谱端进一针DL-苯丙氨酸,那么在液相的色谱图上理论上会出现DL-苯丙氨酸对映体的两个峰,那么当样品流到质谱检测器时,质谱的总离子图上是出现一个峰还是两个峰?液相负责分离,质谱负责定性,质谱可以确定化合物的分子量和分子式,但是可以确定化合物的左右旋对映体么?[/color]
请问在质谱图中为什么会出现负峰?
各位高手请教,岛津2010气相色谱ecd检测器,溶剂怎么会出现双峰? 条件气化室280度,柱箱100度保持2分钟以15度每分钟升到250保持7分钟,检测器温度280,电流1.0 尾吹30.我进了个乙体666,发现溶剂峰出了双峰。
空白,一般包括两种,一种是空白溶液(溶剂);另一种是空白样品溶液,即用制备试样的方法(或生产工艺要求)制备的不含待测样品的溶液。前者是用于考察溶剂对整个检测的影响;而后者多用于考察在制备试样的方法(或生产工艺要求)的前提下,其他因素对检测的影响。如整个处理过程中pH值改变的影响,或前处理过程的影响等等。该问题中的空白指后一种,在实验中为消除色谱分析中某些影响因素的干扰,空白与试样在同样的检测条件下分析,常常用来作为系统适用性(对照)的一个组成部分,也常常用在定量分析中,如在限量检测中有时会带入计算,作为判断样品是否合格的依据。通常空白溶液的谱图有特定的溶剂峰或无峰,然而在某些实验中,空白溶液会出现待测成分的色谱峰,影响实验结果。实验时,如果空白溶液的谱图出现待测成分的色谱峰,会是什么原因造成的呢?
六通阀进样,样品是装置尾气,fid检测器,我想不明白,为什么空柱会有分离效果,求教各位大神。另一根分离柱上每次出现这种情况的时候都是十分钟位置有个峰出现,没定性。[img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808231000006017_1593_3459748_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808231000007557_6942_3459748_3.jpeg[/img]
我在做乳糖酸克拉霉素和烟酸占替诺时都是出现了两个不同位置的主峰,请教高人,为什么一种物质会有两个大峰呢?
顶空进样时,不出峰,使用进样针在空气中抽拉几次会出现一个很高的峰,在吸取样品时也会出现峰,但不确定是否为目标峰,重复性很差,并且目标峰有两个,但是只能出来一个,请问这是为什么,我该怎么处理,谢谢







 我要推广仪器
我要推广仪器
 下载APP
下载APP