[font=宋体][/font][font='微软雅黑','sans-serif'][size=12pt]污染源废水废气比对实验报告中误差保留几位小数或者有效数字,有文件规定吗?[/size][/font][font='微软雅黑','sans-serif'][size=12pt][color=#ff0000]注:问题来自水质分析交流群。[/color][/size][/font][font=宋体][/font]
奈,联苯,菲高效液相分析实验报告 数据处理。。
最近我和另外一个实验室做了一个甜蜜素的对比。但是我们做了2次对比实验误差都很大。 第一次:没有买到甜蜜素标准物质,就找别人要了一点来做实验,结果误差为:33%。 第二次:用自己的标准物质,误差为:16% 在实验中方法都是一样的。仪器不一样:我用的是SC2000型色谱仪,手动进样。另外一个用的是岛津的(型号不清楚)用的是自动进样。 大家帮我分析一下这中间可能造成的原因。
[b][color=#444444]最近我和另外一个实验室做了一个甜蜜素的对比。但是我们做了2次对比实验误差都很大。[/color][color=#444444]第一次:没有买到甜蜜素标准物质,就找别人要了一点来做实验,结果误差为:33%。[/color][color=#444444]第二次:用自己的标准物质,误差为:16%[/color][color=#444444] [/color][color=#444444]在实验中方法都是一样的。仪器不一样:我用的是SC2000型色谱仪,手动进样。另外一个用的是岛津的(型号不清楚)用的是自动进样。[/color][color=#444444] [/color][color=#444444]大家帮我分析一下这中间可能造成的原因。[/color][/b]
气相色谱分析中一般可以将误差分为系统误差、偶然误差和操作误差。系统误差是必然的,也是可以预测的,可以在分析前采取一些措施来消除误差。偶然误差是无法测量和估计的。操作误差是指在分析的过程中一些人为的操作不当或读数失误引起的。气相色谱分析减少误差的方法根据误差产生的原因可以采取合适的减少误差的方法,而误差产生的原因可以从分析流程来看,因此,我们可以在每个环节的操作中选择合适的措施来减少误差。1载气载气是分析的第一步,而气体的纯度会影响到色谱仪的灵敏度,因此,要求气体的纯度在99.999%以上。气体中的杂质会产生基线噪声和鬼峰;气体中的粒状杂质会使得气路控制系统失灵,进而造成分析结果的误差。因此,在气相色谱分析中,首先必须保证气体的纯度,气体需要经过严格的净化才能使用。2进样口进样口的作用就是将样品准确的导入色谱系统中,样品在气化过程中要求不发生任何化学变化,这样才能保证分析结果的准确。而样品在气化中会受到一些因素的影响,主要是隔垫、密封垫和衬管的性能会对样品气化产生影响。第一,隔垫是为了避免外部气体渗入,污染系统,它有效将样品流通与外部空气隔开了。如果隔垫的质量不好,那么就很容易产生鬼峰,也会出现样品的损失、分解等现象。因此,一般选择硅橡胶隔垫,这种隔垫耐高温、气密性好。第二,密封垫将密封色谱柱与色谱系统有效连接起来。密封垫要有良好的密封效果、适宜的内径和耐高温的特点。每更换或维修一次色谱柱,都要更换密封垫;密封垫在使用前必须保持洁净和干燥;密封垫使用15次以上就需要及时更换,确保密封垫的性能。第三,衬管是进样系统的中心元件,样品就是在这里变成气体的。因此,选择衬管时,首先要根据样品的大小来选择合适容积的衬管。其次,如果衬管内壁有活性基团,那么就会对样品的组成产生吸附作用,进而使得色谱仪检测的灵敏度降低。因此,要做好去活工作。最后,根据应用的不同选择不同类型的衬管。合适的衬管可以使得样品最大程度的完全挥发,避免出现热量不均匀的现象,可以减少样品返冲的现象。3取样取样要具有代表性,这样分析结果才具有代表性。因此,取样并不是在某一个部位上直接运用工具取下来的,而是在取不同层次不同部位的样品混合在一起,同时要保证混合的均匀。4进样首先确保进样针的清洁,当针尖存在杂质时,进样的过程中就会使得沉积在壁上的物质在高温气化作用下瞬间发生转移,从而使得分析结果出现一定的误差。所以,在进样时首先确保进样针的清洁,将进样针浸入溶剂中清洁,或是定期对进样针进行清洗。其次是进样量的多少。当进样量过多时,样品在气化过程中就会使得蒸汽体积超过汽化室的容积,然后蒸汽就会到达汽化室的顶部,并在隔垫上出现冷凝现象,蒸汽倒流到载气气路中并在冷的表面产生冷凝现象,这样就造成了样品流失,随后的进样就会出现鬼峰现象,样品分析结果准确性降低。所以,在进样前必须根据衬管的大小选择合适量的样品,并且还要选择合适的气化温度。最后,进样技术。气相色谱分析是一种定量分析方法,因此,分析结果很大程度上依赖于进样的重复性以及操作技术。进样时进样针插入的速度、位置、深度以及操作人员的熟练程度都会影响到分析结果。所以,在进样中,必须根据实际样品的具体情况选择合适的进样技术,选择合适的进样针插入速度、深度、位置等,提高分析结果的准确性。5杜绝人为操作误差在操作过程中,操作人员有时会产生主观误差、过失误差,这样就对分析结果的准确性产生了一定的不良影响。人为误差最常见的就是读数的不准确、取样中贴错标签。为了提高分析结果的准确性,操作人员必须以严谨的态度面对气相色谱分析,在试验前检查各个仪器设备的完好性,进行试验的过程中以高度责任心来认真操作,杜绝操作失误现象的出现;在读数时一定要再三核对,确保读数的准确性,不可随意估测。气相色谱分析是一种定量分析方法,它有效提高了分析的速度,但是在分析过程中任何一个细微的差错都有可能带来分析结果的较大误差。因此,在气相色谱分析过程中,首先要正确认识各个因素对分析结果的影响,正确认识保留时间、待测峰高、样品导入、分流进样系统的影响因素,认识到操作失误对分析结果的影响,从而在试验过程中以严谨认真的态度面对,选择合适的器具和技术方法,一步步消除不良影响,提高分析结果的准确性,减小误差。(来源:互联网)
[font=&]坛子里的各位大牛,公司让我做“HJ 57-2017 固定污染源废气 二氧化硫的测定 定电位电解法 ”的CO干扰实验报告,对于这块不是非常了解,问同事,也说不清楚,还请路过的帮个忙,遇到的问题:[/font][font=&]前提:换的新传感器,标气也是新的。[/font][font=&]1、烟气分析仪测定用配气仪配好SO2和CO的混合标气,CO的浓度怎么都达不到混合的浓度,根据气体浓度不同,相差几十到三四百mg/m3,这个情况是否要进入校准界面调整倍率?[/font][font=&]2、如果每测试一个点,都要去调整倍率,是不是说明这个传感器本身就不准确?每次调整是不是影响上一次调整的倍率?[/font]还请大神解释,不胜感激!
减少[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法在白酒定量分析中误差的方法[b]下载资料网址: http://www.instrument.com.cn/download/search.asp?sel=admin_name&keywords=zjzxwwl2a[/b]无论是毛细管色谱还是填充柱色谱,只要涉及到定量计算就改期存在着一定的误差,怎样才能把误差减少到最低限度以及正确评价定量误差?因此,讨论[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法的定量分析中减少误差的方法十分必要。下面根据内标法定量谈谈实践体会。 一、 取样的代表性 现在大多数产品是中低度酒,由于酒中组分物化特性的影响,致使酒中许多微量成分将分布于不同层次或界面,因此应从酒库取样到色谱室分析的全过程应考虑取混匀后的酒样,如果不注意取样的方式方法,将会给定量工作造成误差。 二、 定量响应因子的准确性 在实际定量工作中,往往引入相对响应因子进行计算,而定量响应因子的准确与否,直接关系到分析结果的可靠程度。若需求得有效的f值,原则上以组份含量相当为依据:一方面,将待测纯组份与纯标准物配成一定比例的混合试样;另一方面以标准样品、混标,专著文献f值等为实际应用f值,必要时可做部分组份的回收实验加以验证后方可使用。 三、 注射器针外壁的清洁 对毛细管柱头进样来说,在进样的过程中沉积在壁上的物质在高温汽化下瞬间发生转移,从而造成定量分析结果的某些偏差,所以在分析不同种类型酒时应严格注意注射器针外壁的清洁。将注射器针浸入溶剂方可达到有效的清洁,也可定期进行清洗。 四、 进样技术的影响 定量分析的精密度与准确度依赖于进样的重复性和操作技术。针对不同规格毛细管柱及特殊的进样方式(柱上进样、分流/不分流进样),对插针的快慢、位置、深度和操作人员的熟练程度以及刻度读数的准确度都有一定的要求,对于大口径柱止进倦毛细管柱,进入柱子的样品量有很好的重现性。对于中口径、细口径分流/不分流进样毛细管柱,当分析的样品组份浓度范围较宽、沸点范围也宽时易产生分流失真,浓度低和沸点高的组份样品回收率低,精密度也差。总之,任何一种进样方法都不能适应所有类型的样品分析,这需要色谱工作者在实际工作中加以选择优化。
无论是毛细管色谱还是填充柱色谱,只要涉及到定量计算就改期存在着一定的误差,怎样才能把误差减少到最低限度以及正确评价定量误差?因此,讨论[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法的定量分析中减少误差的方法十分必要。下面根据内标法定量谈谈实践体会。 一、 取样的代表性 现在大多数产品是中低度酒,由于酒中组分物化特性的影响,致使酒中许多微量成分将分布于不同层次或界面,因此应从酒库取样到色谱室分析的全过程应考虑取混匀后的酒样,如果不注意取样的方式方法,将会给定量工作造成误差。 二、 定量响应因子的准确性 在实际定量工作中,往往引入相对响应因子进行计算,而定量响应因子的准确与否,直接关系到分析结果的可靠程度。若需求得有效的f值,原则上以组份含量相当为依据:一方面,将待测纯组份与纯标准物配成一定比例的混合试样;另一方面以标准样品、混标,专著文献f值等为实际应用f值,必要时可做部分组份的回收实验加以验证后方可使用。 三、 注射器针外壁的清洁 对毛细管柱头进样来说,在进样的过程中沉积在壁上的物质在高温汽化下瞬间发生转移,从而造成定量分析结果的某些偏差,所以在分析不同种类型酒时应严格注意注射器针外壁的清洁。将注射器针浸入溶剂方可达到有效的清洁,也可定期进行清洗。 四、 进样技术的影响 定量分析的精密度与准确度依赖于进样的重复性和操作技术。针对不同规格毛细管柱及特殊的进样方式(柱上进样、分流/不分流进样),对插针的快慢、位置、深度和操作人员的熟练程度以及刻度读数的准确度都有一定的要求,对于大口径柱止进倦毛细管柱,进入柱子的样品量有很好的重现性。对于中口径、细口径分流/不分流进样毛细管柱,当分析的样品组份浓度范围较宽、沸点范围也宽时易产生分流失真,浓度低和沸点高的组份样品回收率低,精密度也差。总之,任何一种进样方法都不能适应所有类型的样品分析,这需要色谱工作者在实际工作中加以选择优化。 五、 硅胶垫的使用周期 硅胶垫的使用频率一般以进样次数作比较,当硅胶垫使用15至20次以上时,应注意及时更换。如果使用国产仪器配套使用的填充柱,应同时擦净内衬管,否则易造成漏气使基线呈台阶、峰型、出现异常等,影响分析结果的可靠性。 六、 进样量的大小 白酒色谱定量使用的内标法,虽然进样量的大小对计算结果无明显影响,但对现行使用的毛细管柱色谱却影响很大。首先,进样量的大小直接影响着分离与定性;第二,进样量的大小直接影响着出峰保留值的变化,造成部分峰保留时间的错位现象,从而影响定量结果,尤其对工作量大、样品较多更不适宜,对于普通填充柱色谱进样量的大、小影响不是太大,但进样量不当也会造成合峰出现,对于毛细管柱来说,这里所谈的进样量与分流比类同。 七、 标样的定期校正 为确保检测数据的可靠性,应定期进行仪器间的相互校正及标样的校验等,从而进一步了解整个色谱系统的运行情况。 八、 怎样正确评价定量误差 1、 单位的一致性 对于填充柱色谱定量的单位通常以mg/100ml,而毛细管色谱定量的单位以mg/100ml计,所以在做分析比较以及评价误差的同时,应考虑单位的一致性。 2、 含量的一致性 无论是标准样品还是色谱纯标样,求f值(响应因子值)时的样品含量与日常分析中酒样含量同样也存在着是否一致的问题。众所周知:含量搞低有不同的误差范围,含量高组份相对的百分误差偏低,含量低组份相对百分误差较高,所以在含量之间的不协调或含量相差悬殊,易造成分析误差的某些偏见,不能对分析结果的误差进行正确的评价。
我们单位在出具化学分析和力学分析试验报告中,都没出具结论(合格还是不合格),我们也曾到外面的实验室做过些外委试验,人家出具的理化报告也没有在报告中出具结论。现在单位要求在所有的试验报告中标明结论(合格还是不合格),对于试验报告该不该出具结论,总是搞不清楚,大家的报告怎么来出的,如果不出具结论这项,有没有依据?
实验报告是对实验观察、比较后所得结果的真实记载,是科学的记录。实验报告的形式可以根据实验内容的不同而分为文字描绘、绘图和列表3种形式。 1.文字描绘:文字描绘是将观察或实验所得结果客观地加以描绘,有时还需要做进一步分析。在此过程中,要抓住主要问题,描绘准确,条理清楚,文字简明。 2.绘图:将所观察标本的形态结构或显微镜下观察视野图通过作图的方式来表达。 (1)准备1支3H和1支HB黑铅笔及橡皮、直尺、绘图纸和削笔刀。绘图必须真实准确,注,意整洁明了。不可抄袭教材或他人的图。(2)图的各部分比例应与标本或图像一致,在绘图纸的一面绘图,每幅图的大小、位置必须分配适宜,布局合理。一般较大的图每页绘1个,较小的图每页绘数个。 (3)图的位置一般偏于纸的左侧,右侧作引线及注字。 (4)绘图时,先用中性铅笔(HB)把标本轮廓及主要部分轻轻绘出,然后添加各部分详细结构,再加以修改,最后用尖的硬铅笔(3H)以清晰的笔画绘出全图。 (5)绘图纸上所有的字必须用硬铅笔以楷书写出,不可潦草。注字引线应水平伸出,各引线不能交叉,图的名称应写在图的下面。3.制图:实验报告中可以用图形表达信息。图形有多种,如曲线图、柱形图、三维图、扇形图等。图形经常表明2种变量(X和Y)之间的关系,2个数轴是相互垂直的。横轴为横坐标(X轴),纵轴为纵坐标(Y轴)。通常,X轴表示自变量(如处理),Y轴表示因变量(如生物效应)。每个数轴都要有说明性的标注和合适的测量单位,每个数轴都要有刻度和参照标记。4.制表:表格通常是简洁、准确、有条理地表示数值型数据的合适方式,它能有效地压缩和展示实验结果,并有助于详尽地对数据进行比较。表格包括的内容如下。 (1)标题:必要时写上参考标注和日期。 (2)行和列的表头:附上合适的测量单位。将相关数据或特性按类别垂直列出,用行展示不同的实验处理、生物类型等。对照值常放在表格的开头,相互比较的列要靠在一起。 (3)数据值:引用有意义的有效数字,根据需要列出统计参数。(4)脚注:解释缩写符号、修饰符号及单个细节。
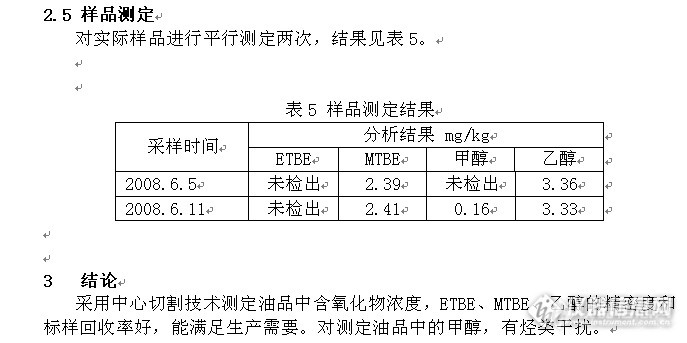
油品含氧化物含量测定法_气相色谱法_试验报告1前言将被测油品用微量注射器注入气相色谱仪,采用中心切割技术 ,使样品从第一根色谱柱DB-1(非极性柱)预分离后的部分馏分,被切割到第二根色谱柱Lowox(极性柱)以进一步进行分离,两根柱子通过特殊的装置连接起来,允许一个或多个感兴趣的组分从第一根柱转移到第二根柱,两根柱间的切换是通过气流控制开关来实现。由于两根柱的极性不同,在第一根柱中共流出的含氧化物组分在第二根柱中得到完全分离。通过氢火焰离子化检测器检测,采用外标法对各含氧化物进行定量。中心切割阀路连接示意图,见图1http://ng1.17img.cn/bbsfiles/images/2014/12/201412201741_528063_2266096_3.png2 实验部分色谱条件:安捷伦气相色谱7890A; 色谱柱1:DB-1非极性柱;30m*0.53mm*2.65um;色谱柱2:Lowox极性柱;10m*0.53mm*10um双检测器:FID 250℃;进样口:200℃色谱柱温度:100℃(5min) 5℃/min 130℃ 10℃/min 225℃(4.5min)2.2 校准曲线绘制称量干净的100mL空容量瓶,加入异辛烷到刻线,称量得出异辛烷的量为70.1781g,依次加入MTBE、ETBE、甲醇、乙醇各约50µL,称量得出各含氧化物的量为:MTBE 0.0374g、ETBE 0.0432g、甲醇 0.0408g、乙醇 0.0391g,计算各含氧化物的浓度为:MTBE 532.9 mg/kg、ETBE 615.5 mg/kg、甲醇 581.3mg/kg、乙醇 557.1mg/kg。对上述溶液进行稀释,得到表1的一系列标准样品。http://ng1.17img.cn/bbsfiles/images/2014/12/201412202019_528085_2266096_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412202021_528086_2266096_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412202021_528087_2266096_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412202021_528088_2266096_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412202021_528089_2266096_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412202025_528092_2266096_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412202025_528093_2266096_3.png
单位刚买了一台万能试验机,求哪位大侠弄一份实验报告的格式,谢谢。我们的试验机是普通型的,再次致谢!

有人说,没逃过课的高中就不算是一个完整的中学生活。人生会留有遗憾。那么,没有手写20几页的实验报告,就不算一个完整的实验过程。人生会留下....后遗症。现在还在手写实验报告吗?晒出我们华丽丽的实验报告吧。参与方式:1. 回复本帖晒出你的实验报告,获得赞的数量达到10个即可获得20积分将努力,获得其它网友点“赞”前10名(且赞总数在20个以上)即可获得可爱造型的可折叠陶瓷果蔬刀 一把(居家旅行必备之物)哦~~2. 微信端参与方式请参见仪器信息网公众号( instrument123)http://ng1.17img.cn/bbsfiles/images/2015/12/201512031737_576222_2961690_3.jpg1“可能是最后一份手写的实验报告?写了三年半,每次最后一页都是“通过这次实验 我学到了很多 提高了动手能力 团结协作能力 把理论结合到实践…”这次的倒还有点儿新意多了几个公式,反正这是我写的最最工整的两页。。”http://ng1.17img.cn/bbsfiles/images/2015/12/201512031727_576215_2961690_3.jpg2(心疼这位写报告的孩纸。。。。繁体字简直要写到手残)什麼年代了,實驗報告還要手寫5頁!五頁耶!!!行行好啊教授工作很多的.....http://ng1.17img.cn/bbsfiles/images/2015/12/201512031728_576216_2961690_3.jpg3老师拿出她学生时代所写的实验报告,惊叹那个时代的一切,整洁的手写卷面,舒服的钢笔字,画得超级逼真的实验仪器。不过才二十年,如今却完全不同。而今天的实验也很成功,克服不耐烦和粗心把蒸馏装置安装好。看着第一滴蒸馏水滴下来,三个人如此兴奋,所谓付出后收获的喜悦无以言表。http://ng1.17img.cn/bbsfiles/images/2015/12/201512031728_576217_2961690_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/12/201512031729_576221_2961690_3.jpg
求一份原子吸收的基础实验报告!做CMA认证,申请检测项目用地~
各位大侠们,上个月考了上岗考试,要写实验报告呢。你说写其他的还好写点吧,让我写噪声。╮(╯▽╰)╭,脑子都憋坏了还是写不出来呀。请问哪位大侠有噪声的实验报告呀,借我参考参考,谢了
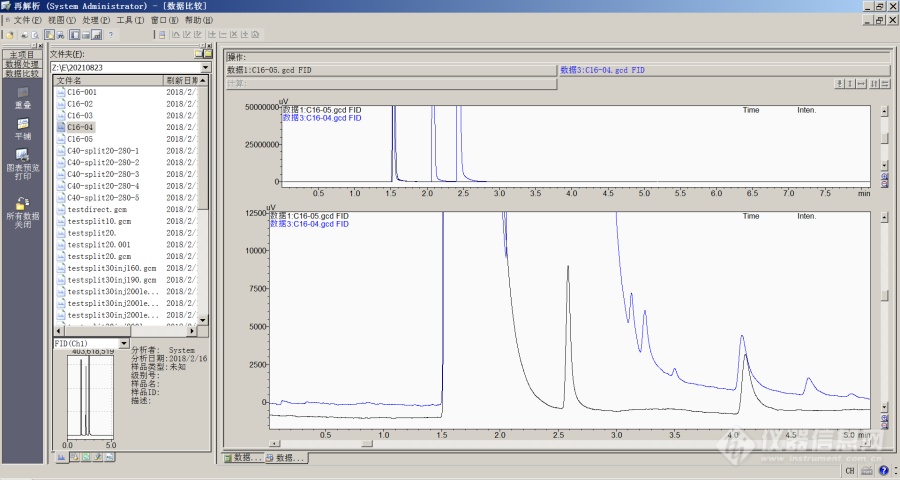
轻溶剂对于目标组分色谱峰影响实验[align=center]概述[/align][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析过程中,在色谱系统内部以气态方式存在的大量溶剂,尤其是小分子、弱保留的轻溶剂,会影响某些目标组分的保留情况。[align=center]情况说明[/align]药品生产和研发行业的用户在开发药品中溶剂残留检测分析方法时,通常会采用两种进样方式——顶空进样和液体直接进样。色谱工作者可能会面临相同的待测物质,在不同进样方式下保留时间不同的问题。可能的情况是,色谱工作者采用直接进样方式实验时,目标组分的保留时间较长;同样的样品改用顶空方式进样实验时,目标组分的保留时间较短。溶剂残留分析过程中经常会使用到分子量较大、保留时间较长的重溶剂。常见分析方法中色谱柱箱的初始温度会比较低,那么在分析的初始阶段,溶剂是呈现为液体状态溶解在固定相中,理论上会影响目标组分的保留。即增加目标物质在“固定相”中的保留,实验的结论是也予以证实。尤其是如果发生重溶剂残留时,目标组分的保留增强现象会变得更加明显。那么轻溶剂会不会影响物质保留呢?为了对这个问题进行探讨,设计了轻溶剂对目标组分影响的实验,选用较为简单的FID计量检定测试样品(100mg/L的正己烷-正十六烷),实验分析条件如下所示:仪器型号: Shimadzu [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]-2010Plus [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]进样口温度: 280℃色谱柱: Rtx-5 30m*0.25mm*0.25um程序升温: 160℃恒温进样量: 1uL进样模式: 分流柱流量: 1ml/min分流比: 40检测器: FID检测器温度: 280℃标准品浓度: 100 mg/L首先使用自动进样器AOC-20i进样正己烷-正十六烷样品,获得数据文件1,正十六烷目标峰保留时间为4.11min。然后再次进样该标准品,进样动作完成后,关闭进样器,迅速在[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]的进样口手工进样2次1uL正己烷,获得数据文件2,正十六烷目标峰保留时间为4.08min。利用Labsolution的数据比较功能,同时打开两个数据进行分析比较,如图1所示:[align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108302300000780_2179_1604036_3.png[/img][/align][align=center]图1 两次进样的数据比较[/align]原始数据文件正十六烷的保留时间和拖尾因子如表1所示;增加溶剂进样之后的数据文件正十六烷的保留时间和拖尾因子如表2所示:表1 第一次进样数据文件的结果表[table][tr][td][align=center]名称[/align][/td][td][align=center]保留时间[/align][/td][td][align=center]拖尾因子[/align][/td][/tr][tr][td][align=center]组分 A[/align][/td][td][align=center]4.11[/align][/td][td][align=center]2.41[/align][/td][/tr][/table]表2 第二次进样数据文件的结果表[table][tr][td][align=center]名称[/align][/td][td][align=center]保留时间[/align][/td][td][align=center]拖尾因子[/align][/td][/tr][tr][td][align=center]组分A[/align][/td][td][align=center]4.08[/align][/td][td][align=center]2.91[/align][/td][/tr][/table]明显可见,当增加轻溶剂进样后,正十六烷的色谱峰保留时间减弱,拖尾程度增加。然后再多次进样原始标样,保留时间基本稳定在4.12min左右。[align=center]原理解释[/align]当载气中溶解有大量轻溶剂之后,目标组分(正十六烷)会在固定相和“含有大量溶剂的载气”之间进行分配,造成分配系数的改变。改变的结果是正十六烷更容易分配于载气中,致使正十六烷的保留减弱,拖尾因子增加。
实验室第一次申请CMA认证,仪器也全是新买的!要做基础实验报告,包括哪些内容?各位用原子吸收举个列子?(方法全是选用国家标准方法的)
[color=#444444]我做一产品,要求喹啉小于0.25%,我们做的是0.1%左右,对方做0.3%左右,在检测方法和色谱柱上可能存在一些差别,但会造成0.2%的误差吗?还有本身小于0.25%要求是否合理呢?[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析的能达到在0.25%以内的误差吗?[/color]
经典的大学物理实验报告参考。供理工科非物理专业使用~~留下你的赞呦!!
当下都在提倡节约用纸,请问各位,你们的试验报告敢用二次纸吗,尤其是对外送顾客的
实验室通过了CNAS-CL01认证,但是由于授权签字人的问题,想咨询一下,不盖CNAS章的实验报告是否可以不用授权签字人进行批准?
[color=#00008B][B]该帖为楼主自己整理,帖中所有楼主撰写的内容未经授权不得转载,否则属侵权违法行为![/B][/color]看到很多版友发帖求助讨论准确性的问题,我特意整理了一个关于定量分析误差问题的帖子,希望对大家有所帮助。高效液相色谱定量分析过程可分为样品的前处理、标准品的配制、进样、色谱分离、检测及数据处理等七个步骤。[B]一 误差的主要来源[/B]随着现在市场销售仪器自动化程度的提高,进样、色谱分离、检测及数据处理等实验环节对实验结果产生的误差越来越小,尤其在高效液相色谱定量分析中,实验结果的误差可能主要来源于样品的前处理及标准品的配制。[B]1、样品的前处理 [/B]样品的萃取率是样品前处理时存在的主要问题。当固-液萃取(含柱分离的前处理方法),液-液萃取时,存在萃取率不高且不稳定的问题。尤其在除去蛋白质时,存在变性蛋白质会吸附一些被测组分,导致萃取率降低的情况。通常,萃取率是通过在式样中添加被测成分在萃取的方法评价的。也就是说,被测成分的增加量和液相色谱中的峰面增大成比例关系,通过这种方法可以确定溶液中被测成分的变化趋势。 如果存在萃取率不稳定的问题,就有必要改变萃取的方法,要预先添加内标,然后萃取。这种情况采用的内标,必须与被测物质的化学结构类似,萃取的萃取率才可能相近。如果回收率不但接近100%而且较为稳定,可以证明这种前处理方法较为可靠。在分析中如果能充分考虑以上误差产生的各种原因,才有可能得到精确的分析结果。 [B]2、标准品的配制[/B]本帖中讨论的问题带有普遍性,影响高效液相色谱分析结果准确性的因素较多,仅在标准溶液的配置过程中,可以分为标准物质的称量,溶液的配制和溶液的储存三个环节。 作为标准溶液使用的标准物质其纯度要求很高,应避免使用纯度不符合要求的试剂,作为标准溶液使用的标准物质具有不可替代性,现在市场有销售的HPLC专用的溶剂及各种标准试剂可供实验选择;其次选择与样品浓度要求相适应的天平,应该尽可能使用高精度的天平,这样才能把由于天平使用带来的称量操作误差降至最低。[B]二 消除误差的方法[/B] 要提高分析结果的准确度,必须考虑在分析过程中可能产生的各种误差,采取有效措施,将这些误差减到最小。[B]1、选择合适的分析方法[/B] 各种分析方法的准确度是不同的。化学分析法对高含量组分的测定能获得准确和较满意的结果,相对误差一般在千分之几。而对低含量组分的测定,化学分析法就达不到这个要求。仪器分析法虽然误差较大,但是由于灵敏度高,可以测出低含量组分。在选择分析方法时,一定要根据组分含量及对准确度的要求,在可能条件下选最佳分析方法。[B]2、增加平行测定的次数[/B] 如前所述增加测定次数可以减少随机误差。在一般分析工作中,测定次数为2—4次。如果没有意外误差发生,基本上可以得到比较准确的分析结果。[B]3、消除测定中的系统误差[/B] 消除测定中系统误差可采取以下措施:其一是做空白实验,即在不加试样的情况下,按试样分析规程在同样操作条件下进行的分析。所得结果的数值称为空白值。然后从试样结果中扣除空白值就得到比较可靠的分析结果。其二是注意仪器校正,具有准确体积的和质量的仪器,如滴定管、移液管、容量瓶和分析天平,都应进行校正,以消除仪器不准所引起的系统误差。因为这些测量数据都是参加分析结果计算的。其三是作对照试验,对照试验就是用同样的分析方法在同样的条件下,用标样代替试样进行的平行测定。将对照试验的测定结果与标样的已知含量相比,其比值称为校正系数。 校正系数=标准试样组分的标准含量/标准试样测定的含量 被测试样的组分含量=测得含量×校正系数 综上所述,在分析过程中检查有无系统误差存在,作对照试验是最有效的办法。通过对照试验可以校正测试结果,消除系统误差。[B]4、样品定量分析过程中的误差[/B]样品处理要尽量减少操作者的技术问题带来的误差,样品的稀释次数、稀释工具都是误差的祸根,应尽量减少稀释次数,稀释工具用高准确度的。样品中的干扰组分会直接影响分析的准确度,而且有些组分会损坏柱子。纯化样品的过程尽量少用蒸发至干的步骤(在色谱分析中这一步又是不可少的),正确操作固相柱萃取、纯化小柱使用的步骤,注意提高每一步的回收率,使用内标法也是一个能准确定量的方法。 在手动进样中进样体积至少是样品定量环管体积的3倍,色谱分离程序要使色谱峰的分离度大于1.5,控制流动相、流量、温度等的平稳。流动相的污染都会抬高基线或减少信噪比,分辨率下降,试验条件的变化,如柱退化、不好的流动相等都能引起保留时间变化,引起一个峰或更多的峰不能被鉴别。正确设定仪器参数,选用合理的数据处理参数,用峰面积计算结果比峰高更精确。[B]三 结论 [/B]以上的分析的是我们能尽量控制的误差,还有一些不是操作者所能控制的误差,如被测定组分易分解、组分的含量高低、介质效应等。我们把能控制的误差减小到最低,那你的结果准确度将更高。
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]系统性试验数据误差的排除方法 使用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],在做原料“冰点”含量测定中,发现系统性试验的一组数据136524、173652、165275、193256,其重复性大于2.0。针对这一问题,采取了以下方法进行分析和排除:一、检查进样过程1、注意进样时间的长短,尽量快速进样。2、用校准过的微量注射器进样。3、每次进样操作应当一致,用同样方式和速度进样。二、检查注温是否平稳1、设定检测器温度200℃2、设定进气口温度180℃3、设定柱温80℃4、检查柱箱控温精度在1;柱箱温度波动小于0.1/h;温度梯度波动小于使用温度的2.三、检查柱的老化程度1、检查基线稳定。2、检查谱峰有无拖尾现象。四、检查FTD检测器拆出检测器喷嘴,发现喷嘴内径变小。这是由于固定液流失,样品在喷嘴燃烧后产生积碳,或使用硅烷化衍生物二氧化硅,污染检测器,检测器线性范围变窗,使灵敏度下降。故卸下喷嘴和收集、清洗:1、用通针通喷嘴,必要时用金相砂纸打磨。2、依次用洗洗剂、水超声清洗。3、在100℃-120℃烘干。通过上述方法处理后,再次进行系统性试验,出现另一组数据:163256、163192、163287、1163112、163232,其重复性合理。希望对大家有用!!!
1主题内容与适用范围在石油加工和自然气净化工艺中产生大量的硫化氢气体,产业上普遍采用克劳斯法处理含有硫化氢的酸性气体,这种方法只有当H2S:SO2为2:1时,才能获得最高转化率。要获得较高的转化率,必须对H2S和SO2进行分析,并且要求分析结果正确可靠。此外,在克劳斯反应过程中还有一些副产物天生,如COS、CS2以及由空气带进的 O2、N2等。为了正确的把握这些组分的含量,对其比率进行最优化控制,减少副反应,进步硫回收率,确定了硫回收过程组成分析的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法。该法已用于硫磺回收过程气中的H2S、SO2、COS、CS2、O2、N2等组分的分析。2仪器与材料[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]。色谱仪应具有下列特性参数。检测器:热导检测器 气化室:要求在本方法使用的温度下能连续运转,为了减少其体积,它和色谱仪的连接间隔应尽量接近 记录仪:积分仪或色谱数据工作站 色谱柱:要匹配双柱,以补偿柱子流失产生的基线漂移 流量控制器:装备有稳流阀和稳压阀,先稳压后稳流 注射器:分格为0.01mL的1mL注射器和100mL注射器。2.2材料担体:分子筛和高分子多孔小球 载气:氢气(纯度≥99.99%) 标准气:H2S、SO2、COS、CS2、O2 稀释气:高纯氮(纯度≥99.99%)。3实验方法为了兼顾硫化物和氧气的分析,选择了如下实验条件。3.1色谱柱和固定相的选择不同的柱材料、柱型、柱长和柱效具有一定的影响。不锈钢柱的优点是不易变形而且具有一定的惰性,用它分析O2、N2、H2S、SO2、COS和CS2等一般化合物是足够稳定的。但是,若使用固定相Chromosorb101时不应使用不锈钢柱。为了进步柱效,色谱柱的外形采用螺旋管,柱内径为 2~3mm,螺旋管直径比色谱柱内径大15~25倍,柱长取2000~3000mm。3.1.2固定相的选择目前,气固色谱中使用的固定相主要有各种形式的碳分子筛和高分子小球等。分子筛是气固色谱中很重要的一种吸附剂,主要用于O2、N2、CO、CH4等永久气体的分离。在使用分子筛前要在真空下350℃左右干燥3h,以便除往水分和其他吸附物。本实验测得了不同温度下2种分子筛的吸水量,如图1所示。当分子中水分在9.37%时,氧和氮已不能完全分开,因此就要活化了,而活化的好坏会直接影响分离效果。分子筛的型号不同性能也不同,实验证实,在相同的条件下,5A分子筛和13X分子筛的保存性能不完全一样,在13X分子筛上 O2的保存时间较短一些,但是5A分子筛的分离能力却比13X分子筛强。高分子多孔小球可用作[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]的固定相,常用的牌号有 Porapak系列,Chrmosorb101、102系列,GDX第列等。GDX是有机芳香族碳氢化合同物,高温下使用体积会收缩,使填充柱产生裂层,在使用前活化会消除此现象,活化温度应高于使用温度20%左右。GDX301是中国自行研制的固定相,主要特点是疏水性强,耐腐蚀、热稳定性好,无固定相流失,可配合高灵敏度检测器使用。通过实验,对分析O2、N2、H2S、SO2、COS和CS2的固定相的种类、色谱柱的长度和材质条件进行了选择优化,结果表明:柱Ⅰ适应于分析H2S、SO2、COS、和CS2 柱Ⅱ适应于分析O2、和N2(见表1)。表1色谱柱及固定相的选定色谱柱柱长柱内径柱材质固定相分离组成编号/mm/mm──────────────────────────────Ⅰ20000.5不锈钢GDX-301H2S、SO2、COS、CS2Ⅱ30000.5不锈钢5A分子筛N2、O2──────────────────────────────3.2色谱柱和检测器温度的控制在50~100℃作了柱温选择实验。当柱温为50℃时,O2、N2分离良好,但H2S、SO2、COS、CS2峰形拖尾较明显,分析时间长,约 15min才能完成分析。当柱温为100℃时,硫化物分析时间可缩短为7min且峰形完整,但O2、N2分离不彻底。结果表明,柱温保持在80℃较适宜。检测器是一种指示并测定载气中被分离组分的装置。实验表明,检测器温度太高会引起噪音的增加,降低检测器的灵敏度,所以,检测器温度控制在100℃较为理想。3.3载气和流速的选择用H2作载气时柱效略低于用氮气作载气,但要缩短分析时间。载气流速对热导检测器的信号值有一定的影响,在热导检测上随载气流速的增加信号即有利于硫化物的分离,又兼顾到O2、N2的分离。3.4桥流在100~600℃mA范围内对桥流作了选择实验,增大桥路电流能明显地增大输出信号 然而桥路电流太大会使基线不稳定,噪音也相应增大。结果表明:采用140mA桥流可满足分析需要。3.5定性及定量分析3.1.5定性分析采用已知纯物质的标准气与未知样品的保存特性进行组分鉴定。即比较已知物和未知物的保存值(保存时间和保存体积),就能确定某一色谱峰代表什么组分。根据保存特性进行鉴定,是最常用的方法。3.5.2定量分析由于各种物质的峰面积与色谱条件有关,和物质的结构和性质没有固定数目关系,因此在进行定量分析时必须要用标准物质进行对比。采用工作曲线定量法或外标法定量。3.6外标气的配制在一个100mL医用注射器内加进少许硬质聚四氟乙烯块,注射器口用一软质橡皮帽封住,先吸取60mL高纯氮,然后用10mL注射器注进一定量的标准气,最后用高纯氮烯释到满刻度,反复摇动,负气体混合均匀。3.7校正曲线的绘制定量进样校正曲线是色谱分析常用的一种简便、快速的尽对定量方法。将已配制好的不同含量标准样,注进[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url],然后将分析结果绘制成峰面积-百分含量校正曲线。在作硫磺回收过程气分析时打进同样体积的分析样品,根据所测得的被分析组分的峰面积值,可直接从外标校正曲线上查出相应组分的百分含量。4灵敏度、误差及最低检测浓度4.1灵敏度目前通常采用迪姆巴特(Dimbat)及其同事提出的方法来测定检测器的灵敏度,即表示为单位浓度的样品所导致检测器的响应值。操纵条件为桥流:140mA 柱温:80℃ 载气:氢气 衰减:1 检测器温度:100℃ 汽化室温度150℃ 载气流速:28mL/min 进样量:1mL。硫化氢为样品,测得:灵敏度S1900mV.mL/mL。4.2正确度表2列出了实验所测得的正确度的具体数据。4.3最低检测浓度在实验条件下实测最低检测浓度见表35结论(1)硫化物分析可采用气固和气液色谱法,但在工艺控制分析中采用气固色谱法具有无固定相流失、柱寿命长等特点,同时,也不会因柱室超温而损坏柱子,可用于高灵敏度检测器。实验证实GDX-301是分析硫化物的理想柱子 (2)由于硫化氢、二氧化硫、二硫化碳和硫氧碳都是极性物质,在分析过程中轻易发生吸附现象而影响分析结果。因此,应尽量缩短采样时间,确保分析结果的正确性 3在分析低含量氧时,应采取有效的密封措施,防止空气中氧的渗透。该方法是为硫磺回收过程气的分析而开发的,方法快速、正确、灵敏,分析误差为±1.4%,试验证实满足硫回收过程气的工艺分析。
CNAS认可实验室出具实验报告费用根据什么来确定呢?请各位帮忙忙,谢谢
[font=Encryption][color=#898989]摘要:[/color][/font][font=Encryption][color=#666666] 原油中烃类[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析的传统方法是先对原油进行族分离得到饱和烃和芳烃,然后分别进行[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析,因此不能反映原油的原始特性和烃类组分的全貌.全二维[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析方法能直接对原油样品进行分析,同时获得饱和烃和芳烃的地球化学参数,避免原油样品经族分离带来的分析误差,缩短分析时间.通过一维、二维色谱柱及升温速率、柱流量、初始柱温等条件实验,优选出原油中烃类全二维[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析的最佳实验条件 应用原油的全二维[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]/飞行时间质谱分析结果,建立了饱和烃、芳烃、萘系列、菲系列及二苯并噻吩化合物的相对位置定性图版,再利用图版对原油中组分进行定性分析 采用面积归一化法进行定量分析,并计算出了石油天然气行业标准所规定的饱和烃与芳烃地球化学参数 精确度实验结果表明,质量分数大于1%的样品多次重复分析的相对偏差小于5%,符合原油实验测定要求.[/color][/font]
[align=center][b][size=24px]气相色谱仪分析的定性依据及定性方法[/size][/b][/align][color=#000000] [size=18px]气相色谱仪[/size][/color][size=18px]的色谱分析包括色谱定性分析和定量分析。今天为大家浅析气相色谱仪的定性分析依据和定性分析方法,仅供色谱工作者参考交流。 (一)气相色谱仪的定性分析依据:气相色谱主要功能不仅是将混合有机物中的各种成分分离开来,而且还要对结果进行定性及定量分析。所谓定性分析就是确定分离出的各组分是什么有机物质,而定量分析就是确定分离组分的量有多少。色谱在定性分析方面远不如其它的有机物结构鉴定技术,但在定量分析方面则远远优于其它的仪器方法。 有机物进入气相色谱后得到两个重要的测试数据:色谱峰保留值和面积,这样气相色谱可根据这两个数据进行定性定量分析。色谱峰保留值是定性分析的依据,而色谱峰面积则是定量分析的依据。 (二)气相色谱仪定性分析方法:气相色谱的定性分析方法主要有保留值定性法、化学[color=#000000]试剂[/color]定性法和检测器定性法。气相色谱的保留值有保留时间和保留体积两种,现在大多数情况下均用保留时间作为保留值。在相同的仪器操作条件和方法下,相同的有机物应有同样的保留时间,即在同一时间出峰。但必须注意:有同样保留时间的有机物并不一定相同。 气相色谱保留时间定性分析方法就是将有机样品组分的保留时间与已知有机物在相同的仪器和操作条件下保留时间相比较,如果两个数值相同或在实验和仪器容许的误差范围之内,就推定未知物组分可能是已知的比较有机物。但是,因为同一有机物在不同的色谱条件和仪器中保留时间有很大的差别,所以用保留时间值对色谱分离组分进行定性只能给初步的判断,绝对多数情况下还需要用其它方法作进一步的确认。一个最常用的确证方法是将可能的有机物加到有机样品中再进行一次气相色谱仪分析,如果有机样品中确含已知有机物的组分,则相应的色谱峰会增大。这样比较两次色谱图峰值的变化,就可以确定前期初步推断是否正确。[/size]
各位大牛,请教下仪器的适用性实验报告具体都要做些什么内容呢,是个什么概念,要怎么做这个报告,求指导。
油中溶解气体色谱分析请问如何分析变压器的油中溶解气体试验报告
[align=center][b][size=24px]白酒分析气相色谱仪分离条件的选择[/size][/b][/align][size=18px] 气相色谱仪分析白酒时,除了选择适合的色谱柱和分析方法外,还要选择好分离的蕞佳操作条件,提高色谱柱的分离效能,增大分离度,获得好的分析结果。色谱技术人员根据实际经验总结出白酒分析气相色谱仪分离条件选择,供大家参考。1. 载气及流速、分流比的选择白酒的气相色谱分析,一般使用FID检测器,常用高纯N2做载气,H2做燃烧气,空气作助燃器。若使用一般填充色谱柱,内径在3~4mm,载气的流量在20~100m L/min。对于内径在0.25mm左右的毛细管色谱柱,载气流量在1~2m L/m in。流速太快会降低色谱柱的分离效能,一般高于蕞佳流速10%左右即可,既保证了色谱柱的分离效能,又能获得比较快的分析速度。H2的流速与载气N2流速相当(毛细管色谱柱载气流量+载气分流的流量),实验证明H2流量∶空气流量=1∶10时,FID检测器蕞灵敏。使用毛细管色谱柱时,分流比的选择直接影响到出峰的个数与分离效果。当分流比为30∶1时蕞为恰当,色谱柱分离效能较高,白酒微量成分分离效果好。载气中微量水分、氢气和空气中的微量杂质对色谱柱和检测器影响很大,严重时会使色谱柱失效,基线不稳,噪声增大,检测器灵敏度下降。所以在载气、H2、空气进入色谱仪之前,应当使用分子筛、硅胶等对气体进行净化处理。2. 色谱柱温的选择白酒中的大部分组分沸点都不高,但沸点范围较宽,为了使低沸点的组分有比较好的分离度,一般初始柱温在50℃。程序升温速度不宜过快,否则分离效果变差,程序升温速度太低,出峰时间长,峰形扁平。一般设定在1~8℃/m in,蕞佳程序升温速度在8℃/m in左右,以保证白酒中各组分在相应的温度下得到良好的分离。蕞终温度不能太高,一般不超过250℃,防止色谱柱温过高,引起固定液挥发流失,分离效能变差,出现基线漂移,或导致色谱柱失效。3. 气化室、检测器温度选择白酒的气相色谱分析中,气化室温度一般高于色谱柱温度50~60℃以上,一般控制在120~200℃,以保证进样时白酒试样中所有的组分都能瞬间变成气体。FID检测器的温度通常控制在150~250℃,避免水蒸汽在检测器中凝结,增大噪声而降低检测器的灵敏度,也可以避免出现检测器点火困难的问题。4. 进样量和进样速度的控制使用填充色谱柱时,柱容量比较大,进样量通常在1~5μL,使用10μL或5μL的微量注射器。采用毛细管色谱柱时,柱容量小,进样量通常在0.1~2μL。进样量低不利于使用低含量组分法进行检测,进样量过高则会导致部分组分峰发生重叠,分离不好。进样速度要求比较快,要求1 s内完成,以保证酒样瞬间气化。如果进样速度太慢,就会引起先插进去的针头部分的酒样先气化,导致色谱峰变宽或者异型,峰形不好,分析误差大的问题。每次进样时,应将微量注射器用被测酒样抽洗5次以上并排净气泡,保证待测试样浓度不发生变化,减少进样带来的误差。5. 其他注意事项为了尽可能地减少分析误差,保证分析结果的准确性,要定期老化色谱柱,在高于使用温度20℃,脱开检测器,通以载气10 h以上,让色谱柱中残留的高沸点组分流出,降低仪器噪声,减小高沸点残余物质的干扰。同时还要定期清理色谱柱头和衬管中积累的不挥发物,防止堵塞色谱柱。每进样50次左右就需更换气化室中的硅橡胶垫,保证气化室不漏气,避免出现色谱峰异常现象。在白酒的气相色谱仪分析中,适当地选择分析方法与测定条件,既可以提高色谱分析的分离效能与检测的灵敏度,又可以提高分析结果的准确度。这就需要我们在实际工作中不断探求与创新,找出每种酒样的蕞佳分析条件,做到准确而快速地分析白酒的微量成分,有效地指导白酒的生产、研发和质量监督,保障白酒的食品安全。[/size]







 我要推广仪器
我要推广仪器
 下载APP
下载APP