[color=#444444]液相色谱使用外标法定量,但是经常出现所有组分含量总和超过100%的现象~~这是为什么,希望高人能够指点一下。[/color]
用高效液相色谱仪外标法测定兽药含量可不可以不建立标准曲线?测出标准品和样品的色谱峰之后直接带入公式求含量可以吗?
同一个样品,检测两次,均采用外标法,一个是气相色谱,一个是液相色谱含量差距很多,气相含量是百分之90左右,液相含量是百分之95左右,求大神分析原因?
高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url](HPLC)中外标法是一种常用的定量分析方法,其基本概念是通过测量已知浓度的标样(外标)和未知浓度的样品的色谱峰面积或峰高,从而计算出样品中目标物质的浓度。一般分为外标单点法和外标曲线法,现在对不同国家的外标法进行对比。 [size=18px][b][color=#595959]01.中国药典中的外标法(2025版的公示稿) [/color][/b][/size][b][size=16px][color=#595959]1)外标法的一般步骤:[/color][/size][size=16px][color=#ff4c41](可以理解为外标法) [/color][/size][/b][size=16px][color=#595959]a.按照各品种项下的规定,精确称取或量取对照品和供试品,配制成溶液; [/color][/size][size=16px][color=#595959]b.分别精确量取对照品溶液和供试品溶液的一定体积,进行色谱分析; [/color][/size][size=16px][color=#595959]c.记录色谱图,并测量待测物质的峰面积(或峰高); [/color][/size][size=16px][color=#595959]d.使用公式 C[sub]X[/sub]=C[sub]R[/sub]×A[sub]X[/sub]/A[sub]R[/sub]计算含量,其中 C[sub]X[/sub]是供试品中待测物质的浓度,C[sub]R[/sub]是对照品的浓度,A[sub]X[/sub]是供试品溶液中待测物质的峰面积(或峰高),A[sub]R[/sub]是对照品溶液中待测物质的峰面积(或峰高)。 [/color][/size][b][size=16px][color=#595959]2)[/color][/size][size=16px][color=#595959]对于成分复杂或待测成分含量变化较大的药物体系,可以采用[/color][/size][size=16px][color=#ff4c41]外标标准曲线法[/color][/size][size=16px][color=#595959]进行含量测定。[/color][/size][/b][size=16px][color=#595959]a.精确称取适量的对照品,或从对照品储备液中精确量取,配制成不同浓度的系列溶液; [/color][/size][size=16px][color=#595959]b.精确量取这些系列溶液,进行色谱分析,记录色谱图,并测量峰响应; [/color][/size][size=16px][color=#595959]c.利用峰响应(或经转换)对浓度绘制标准曲线,并通过最小二乘法计算出回归曲线方程; [/color][/size][size=16px][color=#595959]d.在相同的色谱条件下,精确量取供试品溶液,进行色谱分析,记录色谱图,并测量待测成分的峰响应; e[/color][/size][size=16px][color=#595959].根据待测成分的峰响应(或经转换)和回归曲线方程,确定供试品溶液中待测成分的含量。[/color][/size][size=16px][color=#ff4c41]简单理解就是做一条标准曲线,得出标准曲线方程,再将待测品的峰相应代入方程进行计算,得出样品浓度。 [/color][/size][size=16px][color=#ff4c41][b][color=#595959]02.欧洲药典中的外标法 [/color][/b][/color][/size][color=#ff4c41][b][size=16px][color=#595959]1)[b]多点校准(使用校准函数): [/b][/color][/size][/b][/color][size=16px][color=#595959]a.准备一系列含有不同梯度浓度的对照品溶液,这些浓度应该在已经证明可以得到线性响应的范围内; [/color][/size][size=16px][color=#595959]b.取固定体积的这些对照品溶液进行色谱分析; [/color][/size][size=16px][color=#595959]c.通过色谱图得到峰面积或峰高,并以此绘制校正曲线,横坐标是对照品的量,纵坐标是峰面积或峰高; [/color][/size][size=16px][color=#595959]d.通常通过线性回归得到校准函数; [/color][/size][size=16px][color=#595959]e.按照各论中规定的程序准备待测样品溶液; [/color][/size][size=16px][color=#595959]f.在与制备校准函数相同的操作条件下进行色谱分析; [/color][/size][size=16px][color=#595959]g.测量待测组分的峰面积或峰高,并根据校准函数读取或计算组分的含量; [/color][/size][size=16px][color=#595959]2[b])单点校准: [/b][/color][/size][size=16px][color=#595959]a.在各论中,通常选择一个校准曲线线性范围内的对照品溶液,以及一个浓度接近该对照品溶液的待测样品溶液; [/color][/size][size=16px][color=#595959]b.在固定条件下进行色谱分析,通过比较得到的响应来定量分析组分的含量; [/color][/size][color=#595959]c.在这种方法中,所有操作步骤,如进样程序,都必须在恒定条件下进行 [img=,690,801]https://ng1.17img.cn/bbsfiles/images/2024/09/202409061421193322_2231_3203140_3.jpg!w690x801.jpg[/img] 图1.欧洲药典原文描述[b][color=#595959]03.美国药典中的外标法 [/color][/b][size=16px][color=#595959][b]1)校准函数法(多点校准): [/b][/color][/size][size=16px][color=#595959]a.制备一系列含有不同浓度的对照品标准溶液,这些浓度应在已知能够产生线性响应的范围内; b[/color][/size][size=16px][color=#595959].取固定体积的这些标准溶液进行色谱分析,并注射到色谱系统中; [/color][/size][size=16px][color=#595959]c.通过色谱图得到的峰面积或峰高,绘制校准曲线,横坐标是对照品的量,纵坐标是峰面积或峰高; [/color][/size][size=16px][color=#595959]d.通常通过线性回归方法得到校准函数; [/color][/size][size=16px][color=#595959]e.根据各论中规定的程序制备待测样品溶液; [/color][/size][size=16px][color=#595959]f.在与制备校准函数相同的色谱条件下分析样品,测量待测组分的峰面积或峰高; [/color][/size][size=16px][color=#595959]g.通过校准函数计算出样品中待测组分的含量。[/color][/size][size=16px][color=#595959][b]2)单点校准法:[/b][/color][/size][size=16px][color=#595959]a.在各论中,选择一个浓度在线性范围内的标准溶液,以及一个浓度接近该标准溶液的待测样品溶液; [/color][/size][size=16px][color=#595959]b.在固定条件下进行色谱分析,通过比较得到的响应来定量分析组分的含量; [/color][/size][color=#595959]c.这种方法中,所有操作步骤,包括进样程序,都必须在恒定条件下进行,以确保分析结果的一致性 [img=,690,214]https://ng1.17img.cn/bbsfiles/images/2024/09/202409061424187996_390_3203140_3.jpg!w690x214.jpg[/img] 图2.美国药典原文描述 [color=#595959][b]总结一下:[/b][/color][size=16px]1)中国药典提供了外标法的一般步骤和标准曲线法的详细描述,但没有明确描述单点校准法。 [/size][size=16px]2)欧洲药典和美国药典都详细描述了多点校准和单点校准的方法,并且强调了所有操作步骤需要在恒定条件下进行。(这两家基本是一 致的描述) [/size][size=16px]3)三种药典都强调了在色谱分析中保持操作条件的一致性,以确保分析结果的准确性和重现性 [img=,690,249]https://ng1.17img.cn/bbsfiles/images/2024/09/202409061426098030_8014_3203140_3.jpg!w690x249.jpg[/img] [/size][/color][/color]
请问各位专家:液相色谱外标法测定含量(80%左右)时一般允许的误差是多少呢?2%以内可以吗?
问题:?问一个问题,有一种产品不过滤直接进样,用外标法计算,大学一个月后,计算含量就会偏低,冲一夜后,含量就又高了的,原因是什么呀回复:首先要理解外标法计算的公式原理,再者:样品浓度,峰面积,样品含量之间的关系。
液相色谱法检测含量是液相分析工作的重要的一项,液相色谱法检测含量用的基本是外标法和内标法。其中外标法检测的准确性受到更多因素的影响,更需要丰富的经验来避免或减小误差。本人是做农药制剂分析出身,经过多年的锻炼,自认为对外标法含量检测小有心得,加之最近论坛有坛友问及相关问题,所以想写一篇这方面的小文章,以供大家参考,同时也希望大家补漏拾遗,如有不对的地方,也请指出。首先,我们来看看外标法含量检测的计算公式:含量=m对*P*A样*V样/(m样*A对*V对)*100%其中的各个符号的意义大家都知道,我就不赘述。这其中的任何一项发生了改变,都会使得检测结果发生变化,而这其中的任何一项,都有很多种原因使其发生变化。下面我们就逐一分析。1. m对/m样——对照品称样量/样品称样量1.1 天平准确性天平是否经过校准,其是否在正常工作状态,称量时天平是否带有静电,是否有空气流动,这些都是影响读数的因素。1.2 称量方式采用的是加量法还是减量法,都是很有讲究的。正常情况下,肯定要采用加量法,一次称取到位,这样才能得到准确的称样量。1.3 称量操作称量时,是否足够小心,不会在转移样品时使样品撒漏,这个是非常重要的。1.4 样品性质样品如果具有挥发性,直接称样时,必然会挥发一部分造成误差;另外,如果样品不稳定,或在溶液中不稳定,那么实际上来讲,也是影响了样品量。还有,样品要保证均匀,固体不必说,液体样品要保证是均相,称量之前要摇匀。1.5 称样量对于万分之一天平,称样量最好能在100mg以上;对于十万分之一的天平,称样量应在10mg以上。 2. P——对照品含量2.1 对照品质量对照品是否有合法来源,其含量值是否准确,这个相信大家都知道。2.2 对照品的保存对照品是否按要求保存,也是至关重要的。因为如果对照品没有按照规定保存,那么其右可能会发生降解,进而影响含量值;另外,如果对照品是保存在冰箱中,称量时,应取出放在干燥器中回至室温再称量,否则其很有可能会吸收空气中的水分,影响含量值。同理,如果对照品引湿性很强的话,长期暴露在空气中,也会使其P发生变化。 3. A样/A对——样品峰面积/对照品峰面积3.1 方法的可靠性如果所用的分析方法可靠性不好,样品峰面积或保留时间容易变化,那么检测结果也是很重现性不好的。关于方法的可靠性,做过分析方法验证的人应该都能明白,这里包括系统适用性、精密度、准确性和耐用性以及线性等。3.2 仪器进样精密度这其实可以包括在上述3.1因素中。3.3 残留或污染如果仪器系统被污染产生残留,而本身峰面积就不大的情况下,这个因素产生的误差就更明显了。3.4 积分如果样品峰形不够好,或者分离度不够好的情况下,积分就很重要。 4. V样/V对——样品稀释体积/对照品稀释体积4.1 容量瓶、移液管等体积是否准确一般来说,新买的容量瓶体积是符合要求的,但是也难免会买到个别次品,这就需要进行校验了。4.2 容量瓶、移液管规格这个是很重要的,一般大家都能注意这个问题,做到合理使用。4.3 容量瓶等使用习惯容量瓶在使用时,不能使其至于高于40摄氏度的环境下,否则容量瓶会发生变形。我们以前有个习惯,冬天尤其频繁——瓶子不够用了,而洗过的还没干,就用电吹风吹干,这种行为不光会影响体积,还会使容量瓶瓶口发生变化,摇匀时造成漏液现象。4.4 定量操作这个包括容量瓶和移液管的使用,正确的使用方法就不用说了吧。我只说一些注意事项,一是移液管吸液时,液面不要高刻度太多,放液时千万不要用洗耳球吹,并且尽量保持每次操作时间都一样,粘度大的溶液尤其需要如此。二是观察液面时,要使容量瓶和移液管保持垂直,这时很多人喜欢用三根手指捏住,其实这样不容易保持垂直,最好用两根手指,轻轻捏住就好。三是注意接触容量瓶移液管等的面积越小越好,以免体温加热溶液。4.5 温度[font=Times New Roma
[color=#444444]最近做的不同批次的同一样品,用的是安捷伦1200液相色谱仪,流动相为70%乙腈,波长为215nm。结果如下[/color][color=#444444]1、用面积归一化法计算,纯度为85%,可是用外标法计算含量时,却高达97%(没有乘以总重量,下同)[/color][color=#444444]2、另测一批样品,归一化纯度为86%,外标法计算含量时,却高达100%。[/color][color=#444444]请问这是什么情况?[/color]
大家好,我在用高效液相色谱测芒果苷含量,具体步骤是:取干燥叶片0.1g,加25ML甲醇(萃取剂),称重,超声提取后冷却,用甲醇补足失质量。旋转蒸发仪加热回流,蒸干物用甲醇溶解(这一步是补足失掉甲醇量还是定容到25ML?),最后过滤,滤液体积v1,统一取滤液10ML进行液相色谱测。我只知道液相色谱可以测出芒果苷浓度,但是回归到叶片怎么计算芒果苷含量?哪些体积量是需要用到的。研一新手,实在是搞不懂,恳请大家帮助,谢谢!
求大神列出外标法计算含量的的公式。要具体的啊,按无水干燥物计算

今天接到客户送来的样品,让我们检测一下样品中是否含有杀铃脲,如果含有含量是多少?老规矩先要了解一下杀铃脲,它是一种接触性和胃毒性杀虫剂,属于苯甲酰脲类的昆虫生长调节剂,阻止了昆虫体内合成,使昆虫幼虫虫体畸形而死亡。主要用来防治玉米、棉花、森林、水果和大豆上的鞘翅目、双翅目、鳞翅目害虫,对此类害虫的天敌却无害。此种杀虫药属于低毒杀虫剂,并且效率高,对大多数动物和人无毒害作用,从而被广泛使用。 本次试验将试样用二甲基甲酰胺溶解,通过液相色谱仪,以C18柱为固定相,甲醇+水为流动相,将有效成分与杂质分开,在波长为254nm下,用紫外检测器,数据处理系统,测定流出峰面积,用外标法,定量有效成分的质量分数。一、[b]实验试剂[/b]杀铃脲标准品:已知质量分数为97%;N,N—二甲基甲酰胺:分析纯;水:新蒸二次蒸馏水;甲醇:色谱纯。二、[b]实验仪器[/b]高效液相色谱仪:具有 254nm 波长紫外检测器[img=,375,430]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221113548365_8139_3763765_3.jpg!w690x791.jpg[/img]色谱柱:C18不锈钢柱[img=,242,430]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221115252987_2106_3763765_3.jpg!w690x1226.jpg[/img]微量注射器:50μl。[img=,242,430]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221116089617_8904_3763765_3.jpg!w690x1226.jpg[/img]流动相过滤器[img=,242,430]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221117154197_3452_3763765_3.jpg!w690x1226.jpg[/img]电子天平[img=,297,430]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221118120157_7469_3763765_3.jpg!w690x998.jpg[/img]超声波清洗器[img=,242,430]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221118523747_2538_3763765_3.jpg!w690x1226.jpg[/img]三、[b]色谱条件[/b]流动相:甲醇:水 ﹦80:20流量:1.0ml/min检测波长:254nm进样体积:20μl四、[b]样品前处理[/b]流动相:将甲醇和水按8:2比例混合,经过流动相过滤器过滤后,转移至棕色瓶中,超声后备用。标准品溶液:称取杀铃脲标准品5mg(精确至 0.0002g)于50ml容量瓶中,加入约22ml二甲基甲酰胺溶解,在超声中脱气10min,定容后摇匀。样品溶液:称取样品100mg(精确至 0.0002g)于5ml 容量瓶中,加入约2ml 二甲基甲酰胺溶解,在超声波中脱气 10min,定容后摇匀,用0.5μm孔径过滤器过滤,待测五、[b]测定[/b] 打开机器,设定好上述参数,排气完毕后,待基线走平稳后,使用进样针连续注入三针标准品后,标准品峰面积变化相差小于0.5%,随后按照标准品,样品,样品,标准品的顺序依次进样检测。六、结果及讨论 将测得的两针样品溶液以及样品前后两针标准品溶液的峰面积,分别进行平均,按照下式进行计算: [img=,130,66]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221119280279_4828_3763765_3.png!w130x66.jpg[/img]C样:样品浓度C标:标准品浓度A样:样品平均峰面积A标:标准品平均峰面积[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221123338764_2616_3763765_3.jpg!w690x387.jpg[/img][img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221124134784_89_3763765_3.jpg!w690x387.jpg[/img] 根据谱图判定样品中含有杀铃脲,计算出样品中杀铃脲的含量为:4mg/100mL。
我们开展新的产品的检测方法,1.3环己二酮的含量检测,用乙腈和0.5%乙酸水做流动相(25:75),254波长紫外检测器。用的是单标的外标法检测,取样0.02g,定容50mL,然后稀释后进样的进样浓度(40ppb左右),然后我们测得样品含量的数据很不稳定,前天95-97%左右,昨天99%-103%,今天还是101%左右。求高手指导改进什么地方!!!注:1,单个浓度点的样品的峰面积重现性很好标准偏差0.4%左右。2,我们样品含量应该不超100%,用单标测别家的产品在99%左右。
问题: 问下各位外标法中各峰面积的百分比可以代表含量浓度吗回复: 不可以,纯度和含量明显是两个东西,这个东西是要和标准品进行计算得到的,从峰面积看不出含量的。你先了解下归一化面积法和外标法应用条件吧。
我现在做一糖的液相,岛津的机子,手动进样,用示差折光检测器,以前没对照品只能用面积归一化法,除去两个负峰(有时是一正一负两峰,我是用流动相做溶剂来做的,所以一直不明白这两峰怎么会出现),还有一主峰和一很小的杂质峰,这样计算产品的含量大概能达到99%左右,后来从英国泰莱公司买的对照品,就用外标法做,但计算下来含量只有96%左右,很不明白是怎么回事呢,请大家帮帮我
液相色谱,外标法定量,对标准样品的纯度有要求吗?是不是一定要大于98%?现在只能买到FLUKA质保书上纯度为95%的标准品(GC),可以用于外标法定量吗。大家平时有没有碰到过标准品纯度不高的问题,是不是说明这种物质很难提纯?
请教高效液相色谱仪在进行含量测定时相对偏差为多少?对照品和供试品应各配几份,各进几针平行样?用外标法怎样计算? 谢谢!
本人刚接触液相色谱,想要检测某饮料中某一物质的含量采用外标法,该物质的大体含量不清楚,看了几篇参考文献说外标法中浓度范围最好包括样品浓度,这该怎么做呢?难道扩大外标法中的浓度范围吗,这样好像比较麻烦又不见得准确啊?还有就是进样量应该怎么确定啊?

作者:赵纯玉; 周文杰; 文丽;(广西壮族自治区桂林食品药品检验所;)摘要:目的建立测定醋酸甲羟孕酮片含量的高效液相色谱(HPLC)法。方法色谱柱选用Diamonsil(钻石)C18柱(150mm×4.6mm,5μm),以甲醇-水(65∶35)为流动相,流速为1.0mL/min,紫外检测波长为241nm,柱温为40℃,按外标法以峰面积计算醋酸甲羟孕酮的含量。结果醋酸甲羟孕酮质量浓度在0.5~1000μg/mL范围内与峰面积线性关系良好,r=0.99999(n=8)。该方法与内标法及紫外-可见分光光度法含量测定结果一致,RSD=0.39%(n=3)。结论HPLC法简便易行,准确可靠,重现性好,专属性强。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208271058_386355_1606903_3.jpg
请教:注射用水溶性维生素中关于烟酰胺、等5项的液相检测方法其标准为烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠和核黄素磷酸钠 照高效液相色谱法(中国药典1995年版二部附录Ⅴ D)测定。 色谱条件与系统适用性试验 用氨基键合多孔硅胶为填料,以(0.02mol/L)磷酸二氢钾溶液-乙腈(27:73),用10%盐酸溶液调节pH为5.3的溶液为流动相,流速为1.5ml/min,检测波长:烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠为214nm;核黄素磷酸钠用萤光检测λEX=445nm、λEM=520nm。各组分的分离度应符合要求。 对照品溶液的制备 (1)取烟酰胺对照品约150mg、硝酸硫胺对照品约12mg、盐酸吡哆辛对照品约18mg、泛酸钠对照品约62mg,分别精密称量置50ml量瓶中,加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀,即为对照品溶液(Ⅰ),此溶液置暗处充氮气于零下20℃可保存1个月。(2)取维生素C钠对照品约425mg、核黄素磷酸钠对照品约19mg,精密称定,置50ml量瓶中加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀即为对照品溶液(Ⅱ),此溶液必须临用新鲜配制,并于零下20℃保存,用前放置至室温。 等容混合对照品溶液(Ⅰ)和对照品溶液(Ⅱ)即为对照品溶液。 供试品溶液的制备 取装量差异项下的内容物约2瓶重量,精密称定,置100ml量瓶中,加水溶解并稀释至刻度,摇匀,精密量取15ml置200ml量瓶中,用流动相稀释至刻度。 测定法 取对照品溶液和供试品溶液各10μl,交替注入液相色谱仪,测定,用外标法计算各组分含量,即得。目前存在问题用紫外检测的分不开5种组分,大家有什么好办法,谢谢
有做聚乙氧基化非离子表面活性剂中聚乙二醇含量的测定 高效液相色谱法 的没有,请教下外标法做曲线,聚乙二醇标样600-2000,是只取一种配制不同浓度,做曲线,还是600,1000,2000的都取。具体怎么个情况?
[color=#444444]液相色谱,在不进对照品情况下,如何根据液相图谱面积百分比计算出目标峰的含量?[/color]
[table=100%][tr][td]现在正在测一个样品的含量,有96%的标样一份,待测样品一份 待测样品取了两份 用外标法测含量,结果一份算出来95% 一份96.2% 请问这两份差多少为这个外标实验成功啊?有没有一个标准[/td][/tr][/table]
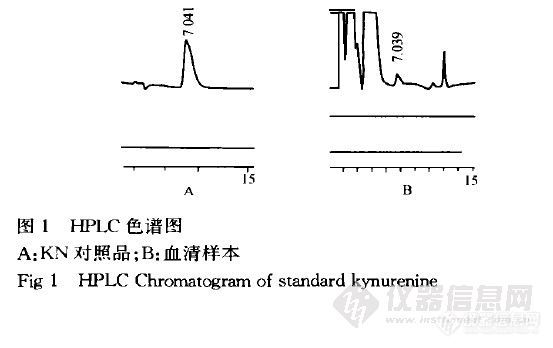
【作者】 陈凤英; 章力行;【机构】 浙江大学医学院附属妇产科医院; 浙江大学医学院附属第二医院 浙江杭州310006; 浙江杭州310009;【摘要】 目的 :建立一种高效液相色谱法测定孕妇血清中的犬尿氨酸 (kynurenine)的含量。 方法 :反相高效液相色谱法 ,采用外标法 ,DiamonsilC1 8柱 ,流动相为甲醇 0 .0 0 1mol·L- 1磷酸二氢钾 (pH =4) (1 0∶90 ) ,检测波长 2 4 5nm。流速 1 .2mL·min- 1 。结果 :kynurenine的线性范围 0 .2 5~ 5 .0 9μmol·L- 1 ,r =0 .9972 ,2 1例孕妇血清Kynurenine的含量为 (0 .98± 0 .2 7) μmol·L- 1 。结论 :本法简便 ,准确 ,适用于临床和科研。 更多还原【关键词】 犬尿氨酸; 高效液相色谱法; 血药浓度; http://ng1.17img.cn/bbsfiles/images/2012/08/201208131445_383518_2352694_3.jpg
有内标物时高效液相色谱法中如何计算含量
液相色谱和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的外标法和内标定量有何区别?是如何处理的?
液相色谱,有结晶水的原料药怎样计算含量我们液相色谱测样品的含量,样品中含两个结晶水,但所用对照品是无水的,计算样品含量时要扣除那两分子的水分吗?
如题,液相色谱中,如何判定计算测的样品中含某一邻苯二甲酸盐的含量?比如,我这个样品测的图谱与标准品对比,并且确定这个峰就是DEHP,那么我应该怎么计算出这个样品中含DEHP的含量是多少?
大家好,请大家来帮助讨论一下外标法测一样品的杂质含量:我现在有一个样品要求用外标法测其含量小于100ppm,现在已经有一杂质的标准品,请问如用外标法测的话如何配制标准杂质的浓度和样品浓度?
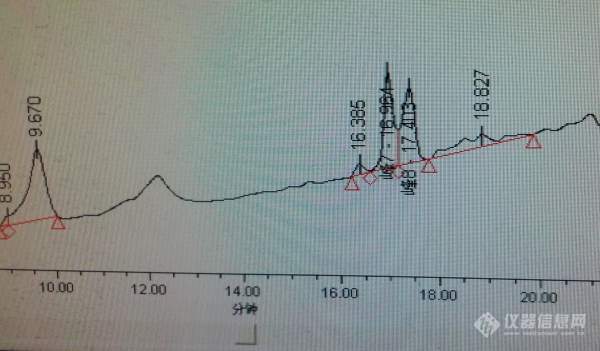
[color=#444444]在使用液相色谱测定环己烷中蒽的含量时,采用的是在文献《有机脱模剂中多环芳烃的高效液相色谱测定》中的方法,温度是20度,检测波长210 nm,采用梯度淋洗方式,开始时,流动相为 (乙腈): (水)=40:60,28 min后,变为V(乙腈):V(水)=82:18,48 min后,变成(乙腈):V(水)=100:0,保持8 min。流速为1ml/min,进样量为10微升。但是这样做走基线之后测出来的图线是飘的,基线偏移很厉害,虽然在17min附近蒽的峰会出现,但是出现的是双峰。我使用的是150mm的C18柱。[/color][color=#444444]师姐说我是因为采用了梯度淋洗的方式所以才会基线偏移,我昨天试了一下直接进V(乙腈):V(水)=70:30的流动相,虽然基线不漂了,但是峰都重叠在一起,全部在7min之前就出来了,我也不知道哪个是蒽,而且峰型都很差。[/color][color=#444444]有没有方法可以在我不改变梯度淋洗的方法的情况下,不出现基线漂移的现象?[/color][color=#444444]或者是有其他更好的流动相进样方法?[/color][color=#444444]我之前出的结果都是双峰,但我不是很想再重做了,这些数据可以用吗?我用的外标法,不知道计算是算两个双峰面积之和还是算平均?[/color][color=#444444][img=,600,351]https://ng1.17img.cn/bbsfiles/images/2019/09/201909231135324988_5836_1849104_3.jpg!w600x351.jpg[/img][/color]
好郁闷 。数据采集后不知道怎么计算样品含量了。按照外表法用峰面积比计算最后结果,含量竞超出100%很多,甚至达到了110%以上。到底什么原因造成计算错误还是怎么样的。以前一直没接触过这方面的东西,现在要自己去做,头都大了。数据采集后,我要计算样品的百分含量,要怎么算呢 ????急啊。我在线等着解答







 我要推广仪器
我要推广仪器
 下载APP
下载APP