液相测多组分时,检测波长是分别取最大吸收波长测,还是建立一个共用的波长,同时测。如果分别测时,不能完全避免另外一个物质出峰,则含量怎么算。谢谢
在用酶联免疫法测定抗原或抗体时,不论是定量试验还是定性试验都要求使用酶标仪进行测定。一般的酶标仪在测定中均有单波长和双波长的模式,并且采用的都是垂直光路。但在日常工作中有时会不太重视单波长和双波长的选择,对使用单、双波长给测定结果带来的较大差异也不很了解,并且实际工作中也出现了使用单波长检测导致抗HCV部分弱阳性的漏检。因此,本人就酶标仪在选择单、双波长使用方面谈谈个人的体会,供同道参考。一 材料和方法1.材料 由上海科华公司提供的乙肝表面抗原测定试剂盒;eppendorf 20-20ul的[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url][/color][/url]。2.仪器 上海科华公司的ST-360酶标仪和BIO-RAD 550酶标仪。3.方法 底物液配制:底物液A、B各5ml混合,加入微量酶标记物,再加入5ml终止液,呈微黄色,混匀待用;利用上海科华公司提供的乙肝表面抗原试剂盒的酶标板,用94孔中准确加入150ul配制好的上述底物液,另2孔中加入150ul已终止的空白底物液作空白。在BIO-RAD 550酶标仪上分别进行450nm单波长和450nm及655nm(无630nm)的双波长检测,在ST-360上分别进行450nm单波长和450nm及630nm的双波长检测吸光度各两次。二 结果 1.分别对所得结果进行统计,发现单、双波长测定结果有较大的差异,双波长测定结果的CV值远小于单波长测定,结果见表1。2.对ST-360两次重复测定结果进行分析,结果基本一致,见表2。表1酶标板吸光度在两台酶标仪上的测定统计结果酶标仪 BIO-RAD550 ST-360 波长 450nm 450nm+655nm 450nm 450nm+630nm 最小值 0.125 0.126 0.130 0.131 最大值 0.154 0.139 0.153 0.141 均值 0.1356 0.1329 0.1399 0.1361 标准差 0.00671 0.00267 0.00467 0.00247 CV(%) 4.94 2.01 3.34 1.82 最大值/最小值 1.232 1.103 1.177 1.076 表2 ST-360酶标仪两次吸光度测定统计结果波长 450mnm 450nm+630nm 1 2 1 2 最小值 0.130 0.129 0.131 0.131 最大值 0.153 0.152 0.141 0.141 均值 0.1399 0.1391 0.1361 0.13711 标准差 0.00467 0.00495 0.00247 0.00229 CV(%) 3.34 3.56 1.82 1.67 三 讨论 1.酶标仪与分光光度计、自动生化分析仪等的吸光度测定有所不同,一般分光光度计是水平光路,而酶标仪则是垂直光路,但测定原理相同,都是使用朗伯-比耳定律,测定的都是样本的吸光度。垂直光的特点是标本吸光度受液体浓缩或稀释的影响小,不足之处是受被测样本液面是否水平、酶标板透光性、孔底是否平整等的影响较大。2.酶标仪在用单波长测定吸光度时,除受到测定干扰(样本的浊度、干扰色等)和电路干扰(包括噪音、漂移、电压等)等因素外,受液体表面张力的影响也很大。在检测过程中,由于液体表面张力的作用,液体的表面不是一个平面,而是形成一个凹面,从侧面看似凹透镜,这样不可避免会影响光路的正常通过。由于凹液面的影响,光线在通过液体时,除正常被液体吸收一部分外,尚有部分被折射和反射(如光线通过凹透镜那样),影响吸光度的检测。而酶标仪使用的又是通过光导纤维传播的点光源,如果每次能在同一部位检测,吸光度的重复性将得到保证,但由于机械运动等造成的误差,不可能保证每孔都在相同部位被检测,因此造成了孔与孔之间有一定的差异。结果见表1,整块板单波长检测的CV在3%以上,吸光度最高值和最低值的相对误差达17%以上。3.在双波长测定中,减少了测定干扰和电路干扰,因此测定结果明显好转,结果见表1,整块板样本的CV在2%以下。ST-360在进行稳定性观察中,如表2所示,两次检测结果基本一致,这表明ST-360的稳定性较好。同时,从表1也可以看到,科华公司生产的ST-360与BIO-RAD 550的检测结果一致,两者的检测性能基本相同。4.由于液体表面张力的不同,导致单波长测定时的误差较大。并且用不同的洗涤剂会影响到最后加入底物和终止液后的液面情况,用加入表面活性剂的洗涤液清洗后,形成的液面更凹,对单波长检测的影响更大,并且与表面活性剂的浓度成正比。而且中性蒸馏水洗涤后,单、双波长检测的结果基本一致。5.在酶联免疫法测定抗原抗体中,由于所使用的底物不论是邻苯二胺(OPD)-H2O2,还是四甲基联苯胺(TMB)-H2O2,显色终止后,在630nm和655nm处的吸光度值都只有吸收峰处(492nm/450nm)吸光度值的1%以下,因此,利用双波长检测,不会影响检测灵敏度。建立在进行酶联免疫检测时,酶标仪比色应该首选双波长。这样可以提高临界值处标本的分析正确度,减少实验误差。
根据酶标仪国家检定规程中,有一项是测酶标仪波长准确度的,请问酶标仪能测波长吗?
测波长一般用什么仪器

[size=16px] 大肠杆菌检测仪如何检测大肠菌群 检测大肠杆菌(Escherichia coli,E. coli)群的方法通常需要使用特定的实验室仪器和技术,而不是简单的手持式检测仪。以下是一般的步骤,说明如何使用传统的实验室方法检测大肠杆菌群: 样品采集: 收集你要检测的样品,这可以是水样、食品样品、环境样品等。 确保采样过程是卫生的,以防止样品污染。 样品预处理: 根据样品类型,可能需要进行样品的预处理步骤。例如,对于食品样品,可能需要将其加入到适当的培养基中以培养细菌。 培养: 将样品或样品预处理液接种到含有营养成分的培养基中,通常使用MacConkey培养基,它有助于选择性生长大肠杆菌。 培养基可能会在37°C的恒温培养箱中培养一段时间,通常为24小时。 大肠杆菌群的鉴定: 观察培养后的培养基,大肠杆菌通常会呈现典型的粉红色或红色粘液样的生长。 使用细菌学技术,例如格拉姆染色、生化测试或分子生物学技术,来进一步确认大肠杆菌的身份。 统计计数: 可以使用传统的计数方法,如涂布法,在含有大肠杆菌的培养基上制作细菌计数板,然后通过计算细菌的数量来确定菌群浓度。 数据分析: 分析实验结果,记录大肠杆菌群的数量,并将其与相关法规或标准进行比较,以确定样品是否符合健康和卫生标准。 需要注意的是,上述步骤是传统的实验室方法,通常需要专业实验室设备和培训的操作人员才能执行。此外,大肠杆菌检测通常需要一定的时间,因为培养和鉴定的过程可能需要一至数天。对于快速的检测需求,也可以使用分子生物学方法,如聚合酶链式反应([url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]),来快速检测大肠杆菌的存在。[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2023/09/202309181131200540_8358_6098850_3.png!w690x690.jpg[/img][/size]
大家好 我是新手我们公司现在想购买一台光谱仪,用来测单色光源的波长误差,我现在只查到了天津拓普的WDS系列,1.推荐一下测单色光源的波长误差的方法2.推荐几款用于测光源波长的仪器,现在网上很多光谱仪都是用来做金属分析用的光谱仪,我想要的是测光源波长的
远红外线的波长用什么仪器测?
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url]器在设置检测波长时发现大于230nm仪器正常,波长改220nm或者210nm,就是小于230nm时,仪器出现提示信息“样品光电二极管上无光”,请教一下这个问题怎么解决?
rt,A300氨基酸分析仪可以双波长同时检测么?还是每次进样只能在一个波长下检测?求前辈帮忙~

[size=18px] 2012年对于分析和实验室仪器行业而言是艰难的一年,在经历了2010年、2011年的强劲增长后,受欧债危机引发新一轮欧美经济低迷的影响也波及到了分析及生命科学仪器行业,因此2012年全球分析和实验室仪器行业增长又跌入了低谷,仅靠新兴市场在各大公司的增长中依旧扮演着重要“角色”,其中中在国市场更是表现突出。虽然各大跨国仪器公司全球业绩不佳,但是在中国他们大多数还都保持了双位数的高增长。2012年赛默飞亚太区收入占公司总收入17%,并且中国区收入逾7亿美元,中国也首次跃居为赛默飞全球第二大市场。对于国内仪器公司,身在中国这个表现强劲的地区,虽然增长也有所放缓,但大多数还是实现了两位数增长。经统计,在已经上市的11家仪器公司中,7家公司2012年增长都在两位数(见下图),其中尤以环境监测为主业的仪器公司2012年业绩普遍表现较好。环境监测仪器行业受环保政策等影响较大,在今后一段时期内还能保持持续增长吗?[/size][img]http://ng1.17img.cn/bbsfiles/images/2013/03/201303311057_433108_1634717_3.jpg[/img]
waters2695-2996这几天在做山柰素的含量测定,同一溶液,昨天测的最大吸收波长是367,今天测的是354,这是怎么原因呢?在这两个波长下同一溶液的含量相差很多。还有就是药典上要求的波长是367,今天测的最大吸收波长是354,差了3个波长。一般规定相差几个波长的误差可以被接受呢?请高手指点啊

同一溶液我用两个波长检测的一个是254nm,一个是235nm,其它条件相同,同一仪器。出来的谱图峰形相似,但是峰面积或者说峰高要差很多,要怎么考虑用哪个波长是对的?
外标法,DAD检测器采用5个波长同时检测,为什么5个波长的定量结果会不一致?难道一定要采用最强吸收波长吗?

多通道波长色散X荧光仪器检测能力相对于顺序型波长色散X荧光仪器,能到达什么水平?比如测定准确度,检测限,精度方面。pb:5ppm,Ti:10pmm ,Ni:10pmm ,V:8ppm ,p:60ppm ,S:12PPM有没有问题。(样品为有机硅)帮朋友调研,欢迎讨论
本人想用液相色谱检测一种物质的纯度,但是文献上没有说在什么波长下检测,试问要是我用紫外光谱仪得到检测波长时,不同溶剂对结果影响大吗?

[size=16px] 大肠杆菌(Escherichia coli,简称E. coli)是一种常见的细菌,其中某些菌株可能会引发食品中的食源性疾病。为了检测食品中的大肠杆菌,通常需要使用专门设计的检测方法和仪器。以下是一般的大肠杆菌检测过程: 样本收集: 首先,需要从待测食品样本中取样。这可能涉及到食品的取样器具和技巧,以确保样本的代表性和卫生。 样品准备: 样品通常需要经过样品制备步骤,以浓缩或净化潜在的大肠杆菌。这可能包括液体培养、离心、过滤或其他处理方法。 培养: 可以将样品接种到培养基中,通过培养大肠杆菌以增殖它们的数量。这通常需要一定的时间,通常在恒温条件下进行。 检测方法选择: 有几种方法可以检测大肠杆菌,包括分子生物学技术、生物化学方法和免疫学方法。以下是一些常见的方法: [url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]检测: 聚合酶链反应([url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url])可以检测DNA中的大肠杆菌基因。这是一种高度敏感和特异性的方法。 生化检测: 这包括检测大肠杆菌的特定代谢产物,如大肠杆菌在培养基中产生的气体或底物转化产物。 免疫学检测: 这些方法使用特定的抗体来检测大肠杆菌的抗原。酶联免疫吸附试验(ELISA)是一种常见的免疫学检测方法。 结果解释: 根据所选的检测方法,可以得出阳性或阴性结果,或者是数量性的结果,反映大肠杆菌在样品中的存在或数量。 结果确认: 有时需要进行进一步的确认测试,以确保结果的准确性。这可能包括对阳性样品进行亚型鉴定,以确定是否存在致病性大肠杆菌株。 数据记录和报告: 检测结果应该被记录并报告给相关部门或机构,以便采取必要的食品安全措施。 云唐大肠杆菌检测仪通常是专门设计用于执行其中一种或多种检测方法的设备,具体取决于实验室或食品工厂的需求。选择适当的检测方法和仪器取决于样品类型、检测的目的以及可用的资源。食品安全是非常重要的,因此确保正确执行大肠杆菌检测是保障公众健康的一项重要措施。[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2023/09/202309200919189229_1894_6098850_3.jpg!w690x690.jpg[/img][/size]
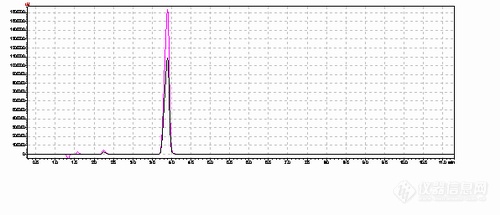
[align=center]液相面积归一法如何确定波长[/align]摘要:一般现在液相检测通常有面积归一法、校正面积归一法、外标法、内标法等几种方法,而检测纯物质,一般只采用面积归一法,而在确定杂质并提取到杂质对照品或提取到主成分对照品时,也可采用加校正因子或不加校正因子的主成分自身对照法进行检测。但是在实际检测过程中,有时候会出现波长确定比较难的情况,本文就如何在使用面积归一法时确定波长并且在主成分波长和杂质波长不同时如何进行选择提出可行性方案。 在使用液相进行检测一种物质的时候,如果没有标准方法、文献可供参考,我们需要通过以下几种方法进行判定波长,第一种是使用DAD二极管阵列检测器,可以很方便的确定被测组分,包括主成分和相关物质的最大吸收波长或者特征波长;第二种是通过紫外可见分光光度计进行波长的扫描,也能得到被测主成分的最大吸收波长或者特征波长,但是无法判定相关杂质的最大吸收波长,而且液相并不一定使用的是最大吸收波长,还可能从分离度等方面进行考虑。第三种是液相紫外检测器有波长扫描功能的,通过扫描波长进行确定目标组分的吸收波长,缺点仍然是只能一个波长测试一遍,才能进行分离度等其他参数的考察。 一般面积归一法测试进行选择波长的过程中,优先选择被测组分的最大吸收波长,其次是特征波长,并且要确定流动相在该波长的吸收不对被测成分产生干扰,选择波长最好比溶剂波长高20nm左右,这样干扰就比较小。但是单从面积归一化法这一个检测方法进行考虑,它是无法判断样品中相应成分的准确含量的。如果合成的相关化合物结构比较类似,都有紫外吸收,其紫外吸收曲线比较一致,有一定的参考价值。但一般情况下,其可靠性是很差的。但是,对于纯物质的判定,往往不能通过外标和内标法进行测定,而是采用面积归一法结合其他方法进行测定。如果出现纯物质的检测,主成分和相关杂质的在一个波长上响应不同,例如:该样品主成分在213和261nm处有两个吸收峰,213nm处为最大吸收峰。213nm测得的纯度较261nm低2%左右,杂质的出峰情况不同。紫外谱图如下:红色为213nm谱图,黑色为261nm谱图[img=,500,215]https://ng1.17img.cn/bbsfiles/images/2019/10/201910240958105464_2581_3295053_3.png!w500x215.jpg[/img][img=,500,312]https://ng1.17img.cn/bbsfiles/images/2019/10/201910240958108954_5224_3295053_3.png!w500x312.jpg[/img] 我们首先可以对主成分及主要杂质进行质谱分析,确定其化学式及其化学性质,如果有该杂质的对照品或者该杂质能够被提取出来,我们可以做杂质与主成分的校正因子,那么在某个波长下,该杂质的相对于主成分的校正因子满足0.8-1.2的范围内,我们就可以确认这个波长是适合的。 如果遇到比较难提取的杂质,但是主成分有相应的对照品,也可以在不同波长下进行主成分对照法进行,配制对照溶液并调节仪器灵敏度后,取供试品溶液和对照溶液适量,分别进样,测量供试品溶液色谱图上各杂质的峰面积,并与对照溶液主成分的峰面积比较,计算杂质含量。通过不同波长下面积归一法与主成分对照法测试的杂质含量的比值进行确定最佳检测波长。如果主成分也没有相应的对照品,这时可以采用衍生化法,不过这种方法仍需了解主成分及相关杂质的化学性质等,然后采用合适的衍生剂,将主要杂质和主成分用衍生的手段把吸收波长调整至非常接近,然后采用相同波长进行检测。这种需要的专业知识较高,而且衍生化法在实际应用中也存在衍生剂干扰、衍生不完全等情况。 还有一种方法是在通过NMR的氢谱、碳谱等相关的谱图先基本确定样品很纯的情况下,通过于其他检测器或者检测方法进行比对,比如我们可以通过卡尔费休法检测水分、顶空色谱检测残留溶剂、灼烧测定灰分、[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]测定一些小分子有机酸,然后结合紫外分光光度计、化学滴定等方法进行主成分的检测,并且与其在液相色谱上的各相关波长结果进行比对,即可得出仪器检测的最佳波长。综上所述,面积归一法并不能只看一个波长的结果,为了得到可靠的结果,我们需要综合判定其在不同波长下的结果进行比对,并根据主成分和杂质的波长响应,得到最佳检测波长,有条件的情况下,还需要通过其他检测器和检测方法进行比较。
检测波长可不可以是一个范围?一定是一个具体的值吗?
有台岛津的CS-9000的双波长飞点薄层扫描仪也不知道准不准,有方法检测吗?
请教一下,为什么国标测总氮要用220,275双波长测试,正常单波长不就可以测了吗?
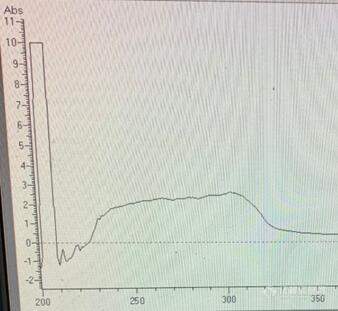
请问在使用液相色谱仪过柱子时,需要设置监测波长和收集波长,我扫了一下紫外光度计,如图所示,请问应该设置监测波长和收集波长多少比较合适?[img=,338,311]https://ng1.17img.cn/bbsfiles/images/2019/05/201905131406077513_7075_1847709_3.jpg!w338x311.jpg[/img]
向大家请教2个问题 1 我在做液相检测的时候 开始出的第一个峰比较正常 后面几个峰出现了负峰的情况。用的是310纳米的检测波长 流动相是 已腈:1/1000 的磷水溶液。 我想问一下一般出现这样的情况是什么问题造成的 (我换234纳米做检测波长就不会出现这样的问题) 2 还有就是检测波长与参比波长到底有什么区别 ,一般我们对于一个物质设置的波长比如上面的 310或者234纳米 到底是检测波长还是参比波长? 原来也问过相似的问题只是回复的不是很清楚希望有经验的朋友能够帮助一下 说的详细点
长输管道敷设施工在完成焊接、防腐、下沟回填作业之后,还要进行管线水压试验。管线试压直接影响管线投产、质量评定竣工验收,管道工程的业主、旋工方、监理方都很关心这道工序。若在试压过程中压力稳不住又难以确定漏点所在处,施工单位通常采用重新分段试压再开挖的方法寻找管线渗漏点,有时要投入很大的力量并需要较长的时间,经济损失也很大。为了有效解决这个问题,运用国产地下[url=http://www.dscr.com.cn/show.asp?id=248]管道防腐层检漏仪[/url]寻找长输管线水压试验的渗漏点,可以取得良好的效果。 (一)发射机的使用 1、发射机接线地点的选择 找出被测管道裸露在地面或可以连接的地方,(一般可以从调压箱,楼前进户管,阀门井,长输管道从测试桩施加信号)取出磁铁,将磁铁与防腐层管道裸露在外的金属部分紧密连接在一起。 找出裸露在外的管道 小锉刀清除表面锈迹 磁铁连接处 *尽量避开多支路的中心点,如计量站,联合站,集输站这些四面八达的管网,会使信号很快的衰减,除掉上面的防腐层和锈迹。 2、发射机接地方式的选择 接地棒一般打在跟管道垂直方向5-10米外的地方,跟管道成垂直方向。 接地棒插入地下 接地线下方的管道感应到很强的信号,会被误判为目标管线,增加探测难度。 3、发射机的连接 取出发射机,连接好输出线,将输出线红色鱼夹连接到磁铁上,将输出线另一端红色\式插头跟接地线上的黑色的式插头连接起来,接地线另一端的黑色鱼夹连接到接地棒上。 4、发射机的调节 打开发射机,观察面板上的参数(功率W)(电压V)(电流MA)(电阻Ω)。通过↑ ↓键调节并查看这些参数,使之阻抗匹配。发射机调好后,便可以进行探测和检漏。 如不匹配工作指示灯发暗或不亮,则需要重新调节,一般发射机的功率,控制在5-10W之间,可根据现场情况来调节发射功率,增大或减小功率。 (二)探管仪的使用 1、探管仪的调节 将探头与探管仪连接好,(未连接不开机),打开接收机 通过↑ ↓键来调节增益的高低。 探杆与探管仪连接 探管仪 当发射机的信号太强,增益已经调到最低信号任然显示1000或1的时候,则必须降低发射机的功率,或通过移动接地棒的位置来解决。 2、 管道定位探测方法 2.1 峰值法:用峰值法(极大值),探头平行于大地,与管线的走向保持垂直探测。 以发射机接线点为圆心,10-20m 为半径做环形探测,边走边转动探头角度,当接收机收到由小变大的信号时,接收机表头数值有小—大—小的变化信号,最大点即为管线位置。 最大值法(示意图) 2.2零值法:选择零值法(极小值)探测时,将探头垂直于大地平面,围绕发射机接线点10m-20m做环形探测时,接收信号将有大—小—大的变化,小点即为管线位置。 3、管道深度探测方式 探管仪测试:一般用45度法。 45度法:管位探到后在正上方做一记号A,将探头转到45度的地方,与管道走向垂直方向移动,当信号最小时再做一记号B,A和B之间的距离即为管到中心到地面的深度。 检漏的方式 常用检测方法:人体电容法 1、 检漏仪的连接 将检漏线跟仪器连接,由两名检测人员各持检漏线一个检漏环,必须与人体紧密接触,保持3-5米 的距离。 检漏线连接处 检漏环跟人体紧密接触 检漏员之间保持3—5米的距离 (注:检漏线绷直尽量不要拖地,检漏员不能穿绝缘鞋) 2、检测仪的调节 打开检漏仪,通过↑ ↓键调节增益,保持表头读数有0-50左右的静态信号。 静态信号在0-50之间 3、检漏的方式 检测时,必须有一个人走在管道正上方,(横向,纵向都可以)。当检漏人员走到破损点处时,检漏仪的声音和表头数值会增大,在漏点的正上方最大。 当破损点较大时,表头读数可能显示为“1”此时应降低增益使显示有读数可比较。 上述检漏方法被称为“人体电容法”,即以人体作为检漏仪的感应元件去寻找发射机发出的信号,正常时信号平稳。当检漏员走到漏点时,由于电流突变,信号也随之变化,喇叭声响和表头指针都有增大显示。为了使漏点处的信号变化更加明显而易于接收和识别,检漏人员在工作中总结以往经验采取了一系列有效的措施。 对检漏仪的操作一定要准确无误,F1-T检漏仪是国内常用的检漏仪器,是长输管道运营单位常备的仪器之一,管道阴级保护人员都能操作。使用地下管道防腐层检漏仪寻找水压试验的渗漏点,首要一条就是操作者必须熟练掌握仪器的操作方法,而且对讯号的判别要有足够的经验。
[color=#444444]HPLC液相色谱,要同时定量测定四种成分,但是其中有三种成分(儿茶素,原儿茶酸,没食子酸)的检测波长相接近大概在280左右,另一种成分(熊果酸)的检测波长大概在220左右,流动相为乙腈和水,单波长不能实现,想用双波长,请问大神们可以用双波长吗?双波长得出的结果图是两个,分析数据的时候怎么分析呀??[/color]
现求购数字万能测长仪JD25-D,请有此货源的朋友与我联系,谢谢!联系人:欧阳小姐联系电话:0760-85833810,13751107849E_mail:mengxun584@163.com
求助:寻找技术专家或者相关单位研制波长色散法测油品和化工产品中低含量的硫的检测仪器,专利或技术转让也可简单描述:我公司希望找人开发波长色散法测油品、化工产品中低含量的硫、氯等元素。如有相关专利或技术也可转让。 技术指标。1、S≤0.1ppm以下 Cl≤0.4ppm以下。 预期经济效益:国外(美国XOS)目前市场价格为70万元人民币左右,日本的同类产品价格为人民币40万元左右,预计研制成功后,年销量大概在50台到100台之间,单位销售价格约为20万元人民币左右,估计销售额1000万~2000万之间,利润大概在300~500万之间。
如图所示,水平放置的两块平行金属板长l =5cm,两板间 距d=1cm,两板间电压为U=90V,且上板带正电,一个电子沿水平方向以速度v0=2.0×107m/s,从两板中央射入。已知电子质量m=9.1×10-31电荷量e=1.6×10-19,求:1、电子偏离金属板的侧位移y0是多少?2、电子飞出电场时的速度是多少?3、电子离开电场后,打在屏上的P点,若s=10cm,求OP的长。
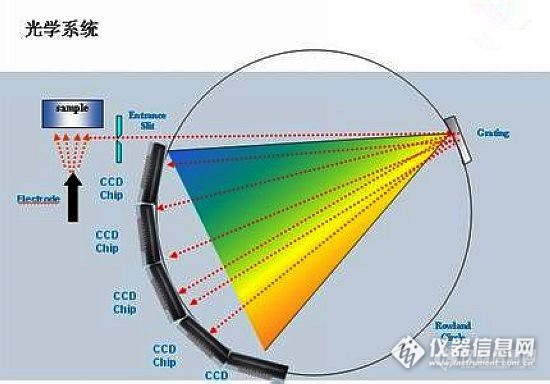
目前一般CCD直读光谱仪大多是采用多个CCD检测器,它们是按元素波长划分的,因为一个CCD检测器很难覆盖整个被测元素的波长范围(严重影响检测限,造成极不均衡),因此用多个CCD检测器分段后,可以较好地解决整个波长范围内兼顾各元素的检测限。http://ng1.17img.cn/bbsfiles/images/2017/01/201701191700_668030_1841897_3.jpg
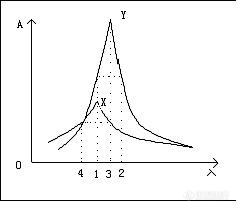
[color=#00008B][B]这几天看到大家对参比波长和检测波长出现比较多的疑问,故整理下帖子,供板油们参考[/B]:[/color]是用紫外可见分光光度计进行全扫描,然后选择最大吸收的波长,如果有几个最大吸收波长的时候,选择杂质峰较少的波长做测定波长。检测波长不是参比波长,只有很少物质测定需要用到参比波长,参比波长主要起到消除噪音的影响选择参比波长的依据是被测样品在参比波长处没有吸收,参比波长应尽可能的接近检测波长。说白了,参比波长就是空白对照的作用如果把参比波长值设成检测波长,样品在紫外光下无吸收。参比波长应设在样品没有吸收的地方,且越靠近样品的检测波长越好。选择参比波长是为了更好的消除噪音的影响。一般参比波长的选择依据是样品在参比波长没有吸收,同时跟检测波长相差不是太大,应尽量靠近,以便消除干扰物质的影响。就象做紫外分光光度计的实验,选择参比溶液一样,为了消除空白溶液中物质的干扰。下面是一个关于参比波长选择的例子:例:X和Y是两种吸光物质的吸收曲线,现采用双波长吸光光度法对它们进行分别测定。如图所示如何选择参比波长及测量波长。(1) 测量X时,Y可视为干扰物质。则选择l1作为测量波长,l2作为参比波长,因为l1和l2两处Y的吸光度相等,可以用以消除Y对X的干扰。 (2) 测量Y时,X可视为干扰物质。则选择l3作为测量波长,l4作为参比波长,因为l3和l4两处X的吸光度相等,可以用以消除X对Y的干扰。 [IMG]http://ng1.17img.cn/bbsfiles/images/2017/01/201701191651_625565_1600062_3.jpg[/IMG]如果你的检测器可以设参比波长,应该是dad或者mwd 检测器。这类检测器的。检测的不是你实际设的波长,比如你设的234,16nm .450,80.表示检测器检到的从226-242nm的紫外吸收图,再平均得到一个数据,参比设置也类似,然后2个相减,就是工作站上显示得到峰高和峰面积。如果你的样品刚好在234nm,吸收最强,你可以设到234,2nm.(资料上一般写频宽宽度设置是最大吸收峰的半峰宽的宽度) 如果你的样品在410-530nm有紫外吸收,你可以把参比设的更高,或者干脆不设参比。
在没有文献的情况下自己找方法时,我通常是采用主峰的最大吸收。现在做杂质检测用的归一法,积分峰面积来检测纯度。后来发现不同的波长(主要有一次忘换波长了),杂质个数没有变化,某个杂质峰增大了很多倍。这样看来的话到底该选择哪个波长下的检测结果?大家那一般情况下怎样选择检测波长呢?







 我要推广仪器
我要推广仪器
 下载APP
下载APP