2011年3月,肯尼亚标准局公布DKS 92-2: 2011“关于合成洗衣粉的机洗要求”和DKS 2292:2011“工业用合成洗衣粉的规范”两项标准草案。 草案规定了主要基于使用烷基芳基磺酸盐的机洗合成洗衣粉和工业用合成洗衣粉的要求和测试方法,该两项草案强调了用于制造洗衣粉的表面活性剂的生物降解能力,并且制定了测定表面活性剂生物降解能力的方法。
一个由瑞士、德国等国家研究人员组成的国际合作研究小组最近制造出了迄今最大的稳定的合成分子PG5。该技术为制造精密分子结构以容纳药物、连接多种物质铺平了道路。 巨型分子PG5直径约10纳米,质量相当于2亿个氢原子,结构好像树枝,大小和烟草花叶病毒相仿。为了制造这种大分子,瑞士联邦理工学院迪尔特·斯格鲁特和同事从标准的聚合反应开始,先把小分子连接起来形成长链,做好了碳氢骨架后,再为其加上由苯环、氮以及碳和氢构成的分枝。经过几次类似的过程,再给每一个分枝加上次级分枝,构成像树一样的结构,就成了PG5。整个过程需要合成17万个分子键。 自然界有很多复杂的大分子,但要在实验室造出这么大的分子却是很困难的,在制造过程中很容易破碎掉。 “合成化学技术迄今还很简单,尚不能达到作为功能单元的尺寸范围。”斯格鲁特说,以前最大的稳定合成分子是聚苯乙烯,质量约为4000万个氢原子。德国马克思·普朗克聚合物研究院的克劳斯·马伦称这项工作为“惊人的”技艺。 斯格鲁特表示,制造骨架和拼接分枝所采用的都是标准技术,他的成功对其他研究人员进一步制造合成大分子是一种激励,以前他们“不够勇敢”去尝试。在应用方面,像PG5这样的分子很适合药物递送,不仅能让药物在各个分枝表面停泊,也能通过分子自身折叠形成空间巢穴。“在装载能力上,目前还没有哪个单体分子能比得上PG5”。
催化合成丁二酸二丁酯第一章 绪论1.1 概要羧酸酯是一类重要的化工原料,低级的酯一般都是水果香味,可作香料(如醋酸异戊酯有香蕉味,戊酸乙酯有苹果香味等)。液态的酯能溶解很多有机物,故常用作溶剂。有些酯还可用作塑料、橡胶的增塑剂。丁二酸二丁酯是一种新型塑料工业的增塑剂,该增塑剂为无色透明液体,常用作有机合成中间体、食物添加剂、气象色谱固定液,是一种昆虫驱避剂,用于驱除蟑螂、蚂蚁等害虫,它的合成与其它酯类化合物一样,由相应的酸和醇通过酯化反应而制得.以往的酯化反应多采用浓硫酸做催化剂,而浓硫酸有腐蚀性,使得酯化反应副反应多、后处理困难、产品色泽较差,同时,在后处理过程中还会产生大量的含硫废水污染环境.为解决浓硫酸作催化剂时的缺点,人们已研究了其它催化剂来代替浓硫酸,但对于丁二酸二丁酯的合成研究的较少,虽有人将TiO2/S042- 固体超强酸用于催化合成丁二酸二丁酯,但该催化剂的制备较为复杂,成本较高,不利于工业化生产.随着人们环保意识的增强,对于酯化反应的催化剂进行了广泛的研究,作者曾注意到结晶硫酸氢钠是一种常见的结晶无机盐,保管、运输、使用均很方便,又能克服无机酸的强腐蚀性,因此作者将研究把硫酸氢钠直接用于催化合成丁二酸二丁酯,主要研究该物质的增塑剂性能和合成该物质所使用的催化剂。
近来很多读者朋友问及我们的“光谱分析网络统计分析管理系统”(LIMS)中的“综合成分稳定度”的概念、意义、用途等方面的问题,现就其作简要说明。注:此帖已在“直读光谱仪”专栏中的“好马如何配好鞍”的回复帖,由于与LIMS系统有关,具有一定的共性,所以也在此与本栏目的朋友们大家共享。1,概念:“综合稳定度”的概念是统计学概率论中的一个纯理论性的概念,是对批量数据对其真值的置信度(可信程度)及其离散性的一个相对量化判断指标。2,意义:“综合稳定度”指标的数值越小,说明置信度越高、离散性越小。反之亦反。但是当该指标小到一定程度(如:等于“0”或接近“0”时,)就会出现实验数据太真实而虚假了。间接的反映了可能是实验设备出现故障,而不能对实验作出真实的检测与分析。针对“直读光谱仪”就可能出现如下故障;氩气问题、真空问题、光路问题、激发台污染等,说明该立即进行设备的检修与保养。3,用途:(对实验室其他“分析仪”系统具有相同用途)---A,对分析试验设备的性能作出判断; 如采用同一标样、相同实验次数(如每台设备作10次)、同一操作者,对不同的设备作试验检测,就可得出设备试验的重复性、置信度、设备完好性等的具体量化判断。---B,对标样的均匀程度作出判断;在同一完好设备下,对一个标样的不同部位(如取十个点位)作十次分析实验,得出的“综合成分稳定度”指标,就可得出该标样均匀程度的---C,对企业产品的质量作出判断;在设备完好的前提下、在某一时段内(如一天、一周、一月等)对企业产品的分析试验批量数据,作“综合成分稳定度”评估,就可间接反映企业生产工艺(炼钢配料、铸造熔炼等工艺)的具体量化判断。---D,对操作者的熟练程度作评价;可以间接反映操作人员的熟练程度、责任心,设备保养周期等实际情况。---E,在试验室信息化管理(LIMS)系统中具有重要的理论与实际意义。
[color=#444444]最近在做3-羟基丁醛的合成,需要用到[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]定量分析产率,没有标样的前提下,用内标法能测出来吗?合成物中含有聚乙醛,丁烯醛,乙酸等副产物,要用什么物质做内标物呢?[/color]
Q1.引物是如何合成的?目前引物合成基本采用固相亚磷酰胺三酯法。DNA合成仪有很多种,无论采用什么机器合成,合成的原理都相同,主要差别在于合成产率的高低,试剂消耗量的不同和单个循环用时的多少。(1) 去保护:加入Deblocking脱去碱基上5'- OH的保护基团DMT,获得游离的5'- OH;(2) 耦合:同时加入活化剂和新的碱基,新的碱基5'-OH仍然被DMT保护,3'端被活化与溶液中游离的5'-OH发生耦合反应;(3) 封闭:耦合反应中极少数5'- OH没有参加反应,用封闭试剂终止其后继续发生反应;(4) 氧化:加入氧化剂使其由核苷亚磷酸酯形成更稳定的核苷磷酸酯。Q2.引物合成后如何处理?切割与脱保护基:将合成好的寡核苷酸链从支持物上化学切割下来。常用新鲜的浓氨水来裂解CPG与初始核苷之间的酯键。断裂下来的寡核苷酸带有自由的3'羟基。纯化:根据所合成寡核苷酸的组成和应用来选定纯化的方法。常用的纯化方法有:C18、OPC、PAGE和HPLC。定量:根据寡核苷酸在260nm处的紫外吸收来定量。储存:分装抽干。Q3.需要什么级别的引物?根据实验需要,确定订购引物的纯度级别。应用引物长度要求纯度级别要求一般PCR扩增60 basePAGE诊断PCR扩增 40baseHEPD, PAGEDNA测序20base左右HEPD亚克隆,点突变等根据实验要求定HEPD, PAGE,HPLC基因构建(全基因合成)根据实验要求定HEPDPAGE反义核酸根据实验要求定HEPDPAGE修饰引物根据实验要求定PAGE, HPLC Q4.引物的质量是不是跟序列有关?四种碱基的性质和各自保护基的性质都有差别。所以合成难度是不一样的。难度最大的当属GC重复多的和序列中还有多个连续的G的引物。尤其对于后者,国内公司一般都做不了20个G以上的引物。实验证明,如果引物中有超过三个连续G的结构,传统方法得到的产物质量就会开始下降。而且目前通用的脱盐、OPC和PAGE方法都无效。鼎国昌盛生物公司拥有的HEPD专利技术能够克服高GC或者高G含量引物的合成和纯化障碍,对普通引物、高GC引物和无论长短的oligo d(G)都能得到同样的非常好的结果。Q5.需要合成多少OD数?根据实验目的确定。一般PCR扩增,2 OD引物,可以做500-1000次50ul标准PCR反应。如果是做基因拼接或退火后做连接,1 OD就足够了。Q6.如何计算引物的浓度?引物保存在高浓度的状况下比较稳定。一般情况下,我们建议将引物的浓度配制成100pmol/ul,称为保存浓度,而引物的工作浓度一般配制成10-50pmol/ul。加水的体积(微升)可直接参照合成报告单上推荐的体积,也可按下列方式计算:V (微升)= OD数x 33 x 10000 /引物的分子量引物的分子量可以从合成报告单上获得。注意:1 OD260= 33 ug/ml。Q7.如何计算引物的Tm值?引物设计软件都可以给出Tm,与引物长度、碱基组成及所使用缓冲夜的离子强度有关。长度为25base以下的引物,Tm计算公式为:Tm = 4℃(G + C)+ 2℃(A + T)对于更长的寡聚核苷酸,Tm计算公式为:Tm = 81.5 + 16.6 x Log10 + 0.41 (GC%) - 600/size**公式中,Size =引物长度。
各位大佬,毕业做乙酸正丁酯的有机合成,要求产率达到80%以上,纯度99.9以上,现在纯度终于做到了,就是产率一直在50%上下左右,跪求各位大佬,疫情原因拖了这么久,马上就六月份了,这可咋办 跪求大佬!!! 下面是我整理的合成细节,大家看看那里不对或者可以做得更好的地方,请大家指出,各位的意见对我很重要谢谢????[img=,690,1226]https://ng1.17img.cn/bbsfiles/images/2020/04/202004132232031655_3401_3490514_3.png[/img][img=,690,1226]https://ng1.17img.cn/bbsfiles/images/2020/04/202004132232037385_8371_3490514_3.png[/img]
请问哪里有卖固相合成仪用于合成多肽的,还有层析缸?谢谢专家!
用9芴酮合成啶酰菌胺,反应中控有那位大神做过,请赐教!
各位老师好,有没有熟练操作希施CS336X多肽合成仪的,我们单位想学习一下这台仪器的使用,南京的最好,方便交流。走过路过的朋友多多发言,或提供学习交流的渠道,非常感谢!
想做“有机电合成香料”,请熟悉电成的朋友谈谈!我有一些基础原料香料,丁香油酚、冬青油、橄榄油、香柏油...想知道需要些什么设备条件、有机电合成香料前沿,谢谢![em23] [em23] [em23] [em23] [em23] [em27] [em27] [em27] [em27] [em27]
我们单位最近开展阴离子合成洗涤剂,在这个实验中遇到问题比较多。第一:分液漏斗质量不好常常会漏、第二:低浓度值结果不稳定、第三:使用GB的方法来做效果不好、第四:实验方法改进之后使用阴离子合成洗涤剂快速检测法。现在求助一下各位站友,有没有什么好的方法来做这个项目。
如题。在研发部门,合成与分析是两大不可或缺的工作角色。但是,合成与分析人员之间的配合以怎样的方式效率比较高,效果比较好呢?身为研发部门的分析人员,总有些许的困惑。请大家来讨论讨论啊。抛砖引玉,先说说我们公司的情况啊。我们的研发部门发展还不是很完善。研发人员有的是一个人做一个项目,有的是几个人共同跟进一个项目。所有的分析检测都由部门的分析室负责。一般的流程是:研发人员在项目开始前,向分析室申报测试项目,由分析室负责人联系标样的购置及分析方法的摸索。项目开始后,交付分析室的分析员分析。但是,这个过程中,总会或多或少的出现一些问题,比如:合成人员抱怨分析人员出报告速度慢,同一个项目的谱图前后对比有差异。而分析人员其实也有自己的难处的,比如:合成人员的路线变化,且未能充分交代样品的信息等等。不知道其他单位是否会出现类似的问题呢?还是说,我们的这种协作方式不够好?听说有些单位是采用一个项目组分配固定的一个或若干个分析人员的方式,这种方式下又会出现哪些问题呢?
请问各位战友,你们在做阴离子合成洗涤剂的时候。有关的器皿是怎么进行洗涤的?我试过 “热水—甲醇—热水” 但是总的来说这么比较费时间,而且玻璃仪器多的时候基本要很长时间搞定!
我想知道关于测试测试丙烯酸酯类的合成单体(如:丙烯酸丁酯,丙烯酸异辛酯之类)使用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]是否可以,如果行,哪选用怎样的色谱仪较好啊????
Q1.引物是如何合成的?目前引物合成基本采用固相亚磷酰胺三酯法。DNA合成仪有很多种,无论采用什么机器合成,合成的原理都相同,主要差别在于合成产率的高低,试剂消耗量的不同和单个循环用时的多少。(1) 去保护:加入Deblocking脱去碱基上5'- OH的保护基团DMT,获得游离的5'- OH;(2) 耦合:同时加入活化剂和新的碱基,新的碱基5'-OH仍然被DMT保护,3'端被活化与溶液中游离的5'-OH发生耦合反应;(3) 封闭:耦合反应中极少数5'- OH没有参加反应,用封闭试剂终止其后继续发生反应;(4) 氧化:加入氧化剂使其由核苷亚磷酸酯形成更稳定的核苷磷酸酯。Q2.引物合成后如何处理?切割与脱保护基:将合成好的寡核苷酸链从支持物上化学切割下来。常用新鲜的浓氨水来裂解CPG与初始核苷之间的酯键。断裂下来的寡核苷酸带有自由的3'羟基。纯化:根据所合成寡核苷酸的组成和应用来选定纯化的方法。常用的纯化方法有:C18、OPC、PAGE和HPLC。定量:根据寡核苷酸在260nm处的紫外吸收来定量。储存:分装抽干。Q3.需要什么级别的引物?根据实验需要,确定订购引物的纯度级别。应用引物长度要求纯度级别要求一般PCR扩增60 basePAGE诊断PCR扩增 40baseHEPD, PAGEDNA测序20base左右HEPD亚克隆,点突变等根据实验要求定HEPD, PAGE,HPLC基因构建(全基因合成)根据实验要求定HEPDPAGE反义核酸根据实验要求定HEPDPAGE修饰引物根据实验要求定PAGE, HPLCQ4.引物的质量是不是跟序列有关?四种碱基的性质和各自保护基的性质都有差别。所以合成难度是不一样的。难度最大的当属GC重复多的和序列中还有多个连续的G的引物。尤其对于后者,国内公司一般都做不了20个G以上的引物。实验证明,如果引物中有超过三个连续G的结构,传统方法得到的产物质量就会开始下降。而且目前通用的脱盐、OPC和PAGE方法都无效。鼎国昌盛生物公司拥有的HEPD专利技术能够克服高GC或者高G含量引物的合成和纯化障碍,对普通引物、高GC引物和无论长短的oligo d(G)都能得到同样的非常好的结果。Q5.需要合成多少OD数?根据实验目的确定。一般PCR扩增,2 OD引物,可以做500-1000次50ul标准PCR反应。如果是做基因拼接或退火后做连接,1 OD就足够了。Q6.如何计算引物的浓度?引物保存在高浓度的状况下比较稳定。一般情况下,我们建议将引物的浓度配制成100pmol/ul,称为保存浓度,而引物的工作浓度一般配制成10-50pmol/ul。加水的体积(微升)可直接参照合成报告单上推荐的体积,也可按下列方式计算:V (微升)= OD数x 33 x 10000 /引物的分子量引物的分子量可以从合成报告单上获得。注意:1 OD260= 33 ug/ml。Q7.如何计算引物的Tm值?引物设计软件都可以给出Tm,与引物长度、碱基组成及所使用缓冲夜的离子强度有关。长度为25base以下的引物,Tm计算公式为:Tm = 4℃(G + C)+ 2℃(A + T)对于更长的寡聚核苷酸,Tm计算公式为:Tm = 81.5 + 16.6 x Log10 + 0.41 (GC%) - 600/size**公式中,Size =引物长度。Q8.引物(含修饰)的分子量是如何确定的?非修饰的引物的分子量(MW)在随引物提供的报告单上可以找到。如果需要估计一个引物的分子量按每个碱基的平均分子量为324.5,引物的分子量=碱基数 x碱基的平均分子量,或按下列公式计算MW= (NA * WA) + (NC * WC) + (NG * WG) + (NT * WT) +(Nmod * Wmod)+(Nx * Wx)+( NI* WI) +16* Ns-62.NA, NG, NC, NT, NI分别为引物中碱基A或G或C或T或I的数量,WA, WG ,WC, WT, WI分别为引物中碱基A或G或C或T或I的分子量,Nmod,Wmod分别为修饰基团的数目和分子量。对于混合碱基的分子量为混合碱基的分子量总合除以混合数,例如G+A混合的分子量为(313.21+329.21)/2 = 321.21。Ns为硫代数目,硫代每个位置增加分子量16。常规碱基分子量BaseMolecular WeightA313.21C289.18G329.21T304.19I314.2U290.17常规修饰基团分子量5’-Biotin405.453’-TAMARA623.605’-(6 FAM)537.463’-Dabsyl498.495’-HEX744.133’-(6 FAM)569.465’-TET675.243’-Amino Modifier C3153.075’-Cy5533.633’-Amino Modifier C7209.185’-Cy3507.593’-Thiol Modifier C3154.12Q9.如何溶解引物?干燥后的引物质地非常疏松,开盖前最好瞬时离心一下,或管垂直向上在桌面上敲敲,将引物粉末收集到管底。根据计算出的体积加入去离子无菌水或TE(pH8.0)缓冲液,室温放置几分钟,振荡助溶,离心将溶液收集到管底。溶解引物用的水一般不要用蒸馏水,因为有些蒸馏水的pH值比较低(pH4-5),引物在这种条件下不稳定。Q10.如何保存引物?引物合成后,经过一系列处理和纯化步骤,旋转干燥而成无色或白色絮状干粉。干 粉:运输时常温运输,-20℃可以保存一年。储存液:配好后分装成几管,避免反复冻融,-20℃可以保存半年。工作液:常规使用,4℃保存,也可-20℃保存,但应避免反复冻融。修饰荧光引物:需要避光保存,尽快使用为宜。Q11.最长可以合成多长的引物?我们合成过100base的引物,但是产率很低。除非需要,建议合成片段长度不要超过80base。引物越长,出现问题的概率就越大,按照目前的引物合成效率,大于80base的粗产品,全长引物的百分比不高,后续处理还有丢失很多,最后的产量很低。Q12.为什么修饰引物的产量要比一般引物低,价格要高?主要因为是修饰单体稳定性较差,偶连时间长,效率低,最后得到的产量自然低于一般的引物。修饰引物通常需要PAGE或HPLC纯化,纯化过程损失大。修饰引物使用的原料是一般引物原料的几百倍,所以产品的价格也高。Q13.引物片段退火后不能连接到载体上是什么问题?连接反应需要引物的5'磷酸基团。如果需要将合成的引物退火直接连接相应的载体上,引物需要磷酸化。磷酸化的产物如果还不能连接载体上,需要检查载体的酶切效果,需要改善引物退火的条件。SiRNA分子具有特殊的对称结构,退火的难度较大,退火时需要提高退火温度。Q14.为什么引物的OD260/OD280小于1.5?需
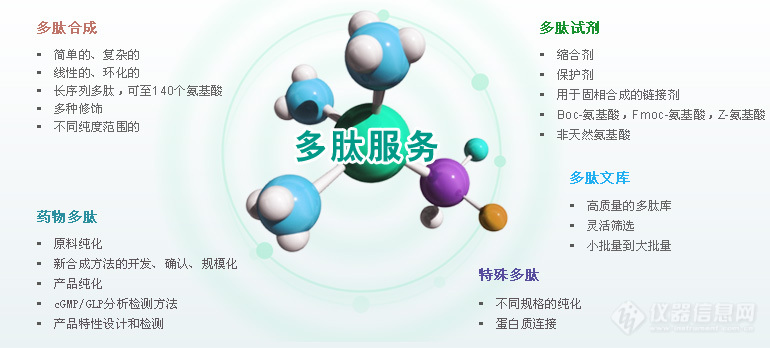
多肽合成又叫肽链合成,是一个固相合成顺序一般从C端(羧基端)向N端(氨基端)合成。过去的多肽合成是在溶液中进行的称为液相合成法。多肽的合成主要分为两条途径:化学合成多肽和生物合成多肽。请移步百度搜“合肥国肽生物”即可多肽合成的原理多肽合成就是如何把各种氨基酸单位按照天然物的氨基酸排列顺序和连接方式连接起来。由于氨基酸在中性条件下是以分子内的两性离子形式(H3+NCH(R)COO-)存在,因此,氨基酸之间直接缩合形成酰胺键的反应在一般条件下是难于进行的。氨基酸酯的反应活性较高。在100℃下加热或者室温下长时间放置都能聚合生成肽酯,但反应并没有定向性,两种氨基酸a1和a2的酯在聚合时将生成a1a2…、a1a1…、a2a1…等各种任意顺序的混合物。为了得到具有特定顺序的合成多肽,采用任意聚合的方法是行不通的,而只能采用逐步缩合的定向多肽合成方法。一般是如下式所示,即先将不需要反应的氨基或羧基用适当的基团暂时保护起来,然后再进行连接反应,以保证多肽合成的定向进行。式中的X和Q分别为氨基和羧基的保护基,它不仅可以防止乱接副反应的发生,还具有能消除氨基酸的两性离子形式,并使之易溶于有机溶剂的作用。Q在有的情况下也可以不是共价连接的基团,而是由有机强碱(如三乙胺)同氨基酸的羧基氢离子组成的有机阳离子。Y为一强的吸电子基团,它能使羧基活化,而有利于另一氨基酸的自由氨基,对其活化羧基的羧基碳原子进行亲核进攻生成酰胺键。由此所得的连接产物是N端和C端都带有保护基的保护肽,要脱去保护基后才能得到自由的肽。如果肽链不是到此为止,而是还需要从N端或C端延长肽链的话,则可以先选择性地脱去X或Q,然后再同新的N保护氨基酸(或肽)或C保护的氨基酸(或肽)进行第二次连接,并依次不断重复下去,直到所需要的肽链长度为止。对于长肽的多肽合成来说,一般有逐步增长和片段缩合两种伸长肽链的方式,前者是由起始的氨基酸(或肽)开始。每连接一次,接长一个氨基酸,后者则是用N保护肽同C保护肽缩合来得到两者长度相加的新的长肽链。对于多肽合成中含有谷氨酸、天冬氨酸、赖氨酸、精氨酸、组氨酸、半胱氨酸等等带侧链功能团的氨基酸的肽来说,为了避免由于侧链功能团所带来的副反应,一般也需要用适当的保护基将侧链基团暂时保护起来。多肽合成方法分类多肽的合成主要分为两条途径:化学合成多肽和生物合成多肽。化学合成主要是以氨基酸与氨基酸之间缩合的形式来进行。在合成含有特定顺序的多肽时,由于多肽合成原料中含有官能度大于2的氨基酸单体,多肽合成时应将不需要反应的基团暂时保护起来,方可进行成肽反应,这样保证了多肽合成目标产物的定向性。多肽的化学合成又分为液相合成和固相合成。多肽液相合成主要分为逐步合成和片段组合两种策略。逐步合成简洁迅速,可用于各种生物活性多肽片段的合成。片段组合法主要包括天然化学连接和施陶丁格连接。近年,多肽液相片段合成法发展迅速,在多肽和蛋白质合成领域已取得了重大突破。在多肽片段合成法中,根据多肽片段的化学特定性或化学选择性,多肽片段能够自发进行连接,得到目标多肽。因为多肽片段含有的氨基酸残基相对较少,所以纯度较高,且易于纯化。多肽的生物合成方法主要包括发酵法、酶解法,随着生物工程技术的发展,以DNA重组技术为主导的基因工程法也被应用于多肽的合成。多肽的固相合成多肽的合成是氨基酸重复添加的过程,通常从C端向N端(氨基端)进行合成。多肽固相合成的原理是将目的肽的第一个氨基酸C端通过共价键与固相载体连接,再以该氨基酸N端为合成起点,经过脱去氨基保护基和过量的已活化的第二个氨基酸进行反应,接长肽链,重复操作,达到理想的合成肽链长度,最后将肽链从树脂上裂解下来,分离纯化,获得目标多肽。1、Boc多肽合成法Boc方法是经典的多肽固相合成法,以Boc作为氨基酸α-氨基的保护基,苄醇类作为侧链保护基,Boc的脱除通常采用三氟乙酸(TFA)进行。多肽合成时将已用Boc保护好的N-α-氨基酸共价交联到树脂上,TFA切除Boc保护基,N端用弱碱中和。肽链的延长通过二环己基碳二亚胺(DCC)活化、偶联进行,最终采用强酸氢氟酸(HF)法或三氟甲磺酸(TFMSA)将合成的目标多肽从树脂上解离。在Boc多肽合成法中,为了便于下一步的多肽合成,反复用酸进行脱保护,一些副反应被带入实验中,例如多肽容易从树脂上切除下来,氨基酸侧链在酸性条件不稳定等。2、Fmoc多肽合成法Carpino和Han以Boc多肽合成法为基础发展起来一种多肽固相合成的新方法——Fmoc多肽合成法。Fmoc多肽合成法以Fmoc作为氨基酸α-氨基的保护基。其优势为在酸性条件下是稳定的,不受TFA等试剂的影响,应用温和的碱处理可脱保护,所以侧链可用易于酸脱除的Boc保护基进行保护。肽段的最后切除可采用TFA/二氯甲烷(DCM)从树脂上定量完成,避免了采用强酸。同时,与Boc法相比,Fmoc法反应条件温和,副反应少,产率高,并且Fmoc基团本身具有特征性紫外吸收,易于监测控制反应的进行。Fmoc法在多肽固相合成领域应用越来越广泛。多肽液相分段合成随着多肽合成的发展,多肽液相分段合成(即多肽片段在溶液中依据其化学专一性或化学选择性,自发连接成长肽的合成方法)在多肽合成领域中的作用越来越突出。其特点在于可以用于长肽的合成,并且纯度高,易于纯化。多肽液相分段合成主要分为天然化学连接和施陶丁格连接。天然化学连接是多肽分段合成的基础方法,局限在于所合成的多肽必须含半光氨酸(Cys)残基,因而限定了天然化学连接方法的应用范围。天然化学连接方法的延伸包括化学区域选择连接、可除去辅助基连接、光敏感辅助基连接。施陶丁格连接方法是另一种基础的片段连接方法,其为多肽片段连接途径开拓了更广阔的思路。正交化学连接方法是施陶丁格连接方法的延伸,通过简化膦硫酯辅助基来提高片段间的缩合率。其他多肽合成方法1、氨基酸的羧内酸酐法(NCA)氨基酸的羧内酸酐的氨基保护基也可活化羧基。NCA的原理:在碱性条件下,氨基酸阴离子与NCA形成一个更稳定的氨基甲酸酯类离子,在酸化时该离子失去二氧化碳,生成二肽。生成的二肽又与其他的NCA结合,反复进行。NCA适用于短链肽片段的多肽合成,其周期短、操作简单、成本低、得到产物分子量高,在目前多肽合成中所占比例较大,技术也较为通用。2、组合化学法20世纪80年代,以固相多肽合成为基础提出了组合化学法,即氨基酸的构建单元通过组合的方式进行连接,合成出含有大量化合物的化学库,并从中筛选出具有某种理化性质或药理活性化合物的一套多肽合成策略和筛选方案。组合化学法的多肽合成策略主要包括:混合-均分法、迭代法、光控定位组合库法、茶叶袋法等。组合化学法的最大优点在于可同时合成多种化合物,并且能最大限度地筛选各种新化合物及其异构体。3、酶解法酶解法是用生物酶降解植物蛋白质和动物蛋白质,获得小分子多肽。酶解法因其多肽产量低、投资大、周期长、污染严重,未能实现工业化生产。酶解法获得的多肽能够保留蛋白质原有的营养价值,并且可以获得比原蛋白质更多的功能,更加绿色,更加健康。4、基因工程法基因工程法主要以DNA重组技术为基础,通过合适的DNA模板来控制多肽的序列合成。有研究者通过基因工程法获得了准弹性蛋白-聚缬氨酸-脯氨酸-甘氨酸-缬氨酸-甘氨酸肽(VPGVG)。利用基因工程技术生产的活性多肽还有肽类抗生素、干扰素类、白介素类、生长因子类、肿瘤坏死因子、人生长激素,血液中凝血因子、促红细胞生成素,组织非蛋白纤溶酶原等。基因工程法合成多肽具有表达定向性强,安全卫生,原料来源广泛和成本低等优点,但因存在高效表达,不易分离,产率低的问题,难以实现规模化生产。5、发酵法发酵法是从微生物代谢产物中获得多肽的方法。虽然发酵法的成本低,但其应用范围较窄,因为现在微生物能够独立合成的聚氨基酸只有ε-聚赖氨酸(ε-PL)、γ-聚谷氨酸(γ-PGA)和蓝细菌肽。[align=center][img=,770,348]https://ng1.17img.cn/bbsfiles/images/2019/03/201903151633244062_8177_3531468_3.jpg!w770x348.jpg[/img][/align]请移步百度搜“合肥国肽生物”即可我们主要提供:多肽合成、定制多肽、同位素标记肽、人工胰岛素、磷酸肽、生物素标记肽、荧光标记肽(Cy3、Cy5、Fitc、AMC等)、目录肽、偶联蛋白(KLH、BSA、OVA等)、化妆品肽、多肽文库构建、抗体服务、糖肽、订书肽、药物肽、RGD环肽等。
采用生物质气化制取的合成气,气化炉中高温1000度左右,压力为负压 -99.8KPa,粗合成气中含有焦油、粉尘和水。 (1)怎样在高温负压的气化炉中取样?采用在线分析还是取样后离线分析? (2)如何测粗合成气中的焦油、粉尘和水含量?采用什么仪器?测试原理是什么? (3)粗合成气进入气象色谱分析前,如何去除粗合成气中的焦油、粉尘和水?气相色谱需要什么配置? 大家能否推荐一下仪器的厂家?谢谢。
阴离子合成洗涤剂可以用第一个分液漏斗加5ml萃取3次,三次合并于第二个放25ml洗涤液的分液漏斗中,萃取,萃取液放比色管,再用5ml三氯甲烷萃取洗涤液两次,合并比色管,定容比色。
我现在需要对含有微量乙烯的合成气进行GC分析,有哪位高手有这方面的经验或资料,请指导一下吧。谢谢 此待分析的合成气组分大概是:氢气37%,一氧化碳34%,乙烯0.2%,和其他。 最好能够提供较详细的色谱方法和资料,非常感谢
1.多肽合成的基本原理?多肽固相合成法是多肽合成化學的一個重大的突破。它的最大特點是不必純化中間產物,合成過程可以連續進行,進而為多肽合成的自動化奠定了基礎。目前全自動多肽的合成,基本都是固相合成。其基本過程如下:基於Fmoc化學合成,先將所要合成的目標多肽的C-端氨基酸的羧基以共價鍵形式與一個不溶性的高分子樹脂相連,然後以這一氨基酸的氨基作為多肽合成的起點,同其他的氨基酸已經活化的羧基作用形成肽鍵,不斷重複這一過程,即可得到多肽。根據多肽的氨基酸組成不同,多肽後處理方式不同,純化方式也有差異。2.做免疫用的多肽多長為合適?答:一般約10-15個氨基酸,當然長一些免疫效果好一些,不過合成費用也會增加。MAP多肽則希望長度在15aa以上,效果較好。另外,10aa以下的多肽免疫效果比較差。3.免疫用多肽的純度需要很高嗎?答:一般而言, 免疫用Peptide,70-85%即可。4.我們合成的多肽溶解性不好,多肽就有問題對嗎?答:很難準確預測一個多肽的溶解性及合適的溶劑是什麼。如果多肽難以溶解就認為多肽合成有問題這個觀念並不正確。5.多肽狀態是如何?如何保存儲存?答:我們提供的多肽是粉末狀,一般為灰白色,組成不同,多肽粉末的顏色有差異,多肽一般長期保存需要避光保存,並應保存在-20度,短期可以保存在4度。可以短時間的話是以室溫運輸。6.如何溶解多肽?答:溶解多肽是非常複雜的事情,一般很難一下子確定合適的溶劑。通常是先取一點試驗,在沒有確定合適的溶劑前千萬不要合部溶解。下列方法有助於您選擇合適的溶劑:(1)判定多肽的電荷特定,設定酸性氨基酸Asp(D),Glu(E)和C端COOH為-1;鹼性氨基酸Lys(K),Arg(R),His(H)及N端NH2為+1,其他氨基酸的電荷為0。計算出將電荷數。(2)如果淨電荷數 0,多肽為鹼性,用水溶解:如果不溶解或溶解性不大,加入醋酸(10%以上);如果多肽還不能溶解,加入少量TFA(25ul)溶解,然後加入500ul水稀釋。(3)如果淨電荷數0,多肽為酸性,用水溶解;如果不溶解或溶解性不大,加入氨水(25ul)溶解,然後加入500ul水稀釋。(4)如果淨電荷數=0,多肽為中性,一般需要用有機溶劑如乙腈,甲醇或異丙醇,DMSO等溶解。還有人建議需要尿素來溶解疏水性很大的多肽。7.非HPLC純化的多肽中有哪些雜質?答:粗品和脫鹽級別的多肽中多肽和非多肽類雜質:如非全長多肽和多肽後處理的一些原料如DTT、TFA等8.HPLC純化的多肽有哪些雜質?答:經過HPLC純化的多肽,仍會有一些一些雜質存在,其中的雜質主要是短肽和微量TFA。9.多長的多肽為合適?答:多肽合成需要考慮多肽的長度,電荷,親疏水性等因素。長度越長,合成粗品的純度和產率都隨著降低,純化的難度和無法合成的幾率就會大些。當然多肽功能區的序列是無法改變的,但是為了多肽的順利合成,有時不得不在功能取的上下游增加一些輔助氨基酸,以改善多肽的溶解性和親疏水性。如果多肽太短,合成也可能有問題,主要問題是合成的多肽在後處理過程中有一定的難度,5肽以下的多肽,一般要有疏水的氨基酸,否則後處理難度加大。15個氨基酸殘基以下的多肽一般都可以得到滿意的產率和得率。10.如何從多肽序列中判定多肽的溶解性?(1)多肽中如果含有高比例的疏水性很強的氨基酸如和Leu,Val,IIe,Met,Phe和Trp,多肽很難溶解與水性溶液中或根本不可能溶解。這些氨基酸無論是純化或合成,都有可能有問題。(2)一般情況下疏水性氨基酸的比例50%,不能連續5個連續aa為疏水性,帶電荷的氨基酸的(正電荷K,R,H,N-terminus,負電荷D,E,C- terminus)的比例達到20%,在多肽的N或C短如果能增加極性氨基酸,也可以改善溶解性。11.為什麼含有Cys,Met,或Trp的多肽難合成?答:含有Cys, Met,或Trp的多肽難以合成,同時難以獲得高純度的產品。主要因為這些基團不穩定,易氧化。這些多肽的使用和儲存都需要特別注意,避免反復開啟蓋子。12.為什麼有些多肽的合成產率或純度會比較低?答:多肽合成與引子合成有比較大的區別,不能合成的引子很少,但是不能合成的多肽經常有。如Val,Ile,Tyr,Phe,Trp,Leu,Gln,和Thr這些氨基酸比鄰或重複時,多肽鏈在合成過程中不能完全舒展溶解,合成效率下降。以下幾種情形,合成效率和產物的純度都比較低,如:重複Pro,Ser-Ser,重複Asp,4個連續Gly等.13.多肽是如何純化的?答:多肽純化一般使用反相柱(如C8,C18等),214nm。緩衝體系通常為含TFA的溶劑,pH 2.0 。Buffer A為含0.1%TFA in ddH2O,Buffer B為1%TFA/ACN/ pH 2.0。純化前用Buffer A溶解;如果溶解不好,用Buffer B溶解後,然後用Buffer A稀釋;對疏水性強的多肽,有時還需要加入少量的Formic Acid或醋酸。HPLC分析多肽粗產物,如果多肽不長(15aa以下),一般會有主峰,主峰通常為全長產物;對於20aa以上的長肽,如果沒有主峰,HPLC需搭配Mass來判定分子量,進而確定哪個峰是所要合成的多肽。

有机合成大致分为两方面:①基本有机合成。包括从煤炭、石油、水和空气等原材料合成重要化学工业原料,如合成纤维、塑料和合成橡胶的原料,溶剂,增塑剂,汽油等,其产量几乎接近于钢铁的数量级。②精细有机合成。包括从较简单的原料合成较复杂分子的化合物,如化学试剂、医药、农药、染料、香料和洗涤剂等。在微波的条件下,利用其加热快速、均质与选择性等优点,应用于现代有机合成研究中的技术,称为微波有机合成。微波有机合成特点:(a)加热速度快。由于微波能够深入物质的内部,而不是依靠物质本身的热传导,因此只需要常规方法十分之一到百分之一的时间就可完成整个加热过程。(b)热能利用率高,节省能源,无公害,有利于改善劳动条件。(c)反应灵敏。常规的加热方法不论是电热、蒸汽、热空气等,要达到一定的温度都需要一段时间,而利用微波加热,调整微波输出功率,物质加热情况立即无惰性地随着改变,这样便于自动化控制。(d)产品质量高。微波加热温度均匀,表里一致,对于外形复杂的物体,其加热均匀性也比其它加热方法好。对于有的物质还可以产生一些有利的物理或化学作用。影响微波有机反应的因素:从一个反应体系来看,我们通常可以改变的是:溶剂,反应物,催化剂,以及反应体系中各物质的比例。从辅助条件看:搅拌强度,是否预搅拌,气体保护,冷凝回流,反应压力等。从微波反应看:反应温度,反应时间,微波功率等。微波在有机合成上的效应:1, 热效应(包含由温度变化带来的特殊效应),大家比较了解,不多说。2, 催化诱导效应,许多有机化合物都不直接明显地吸收微波,但可利用某种强烈吸收微波的“敏化剂”把微波能传给这些物质而诱发化学反应,这一概念已被用作诱发和控制催化反应的依据。如果选用这种“敏化剂”作催化剂或催化剂的载体,就可在微波辐照下实现某些催化反应,这就是所谓的微波诱导催化。3, 微波的非热效应,非热效应则是一种无法用温度变化来解释的特殊效应,微波作用于化学反应,改变了反应的动力学,改变了反应的活化能和指前因子,使反应更容易进行。微波对化学反应增强有选择性加热的热点效应、分子搅拌、化学键振动以及微波增强物质扩散等作用。微波有机合成注意事项:一般在微波场能发生爆炸的反应是不能做的,比如(反应是连锁反应,反应物和溶剂带有CN或者N基团),有些反应要小心,比如放热反应或产生气体的反应。微波反应的温度我们通常设定在沸点+10℃,沸点+25℃,沸点+50℃以内。防止反应压力过大。我们也必须留意,某些溶剂在微波场作用下会产生分解的情况,DMSO在120℃下,30分钟开始分解,DMF在150℃下,30分钟开始分解。这些情况需要避免发生。新仪公司最新推出的UW-2000微波合成萃取仪适用于常压及加压有机合成。同时可加超声波和紫外线辐射,兼具人性化的软件设计,对广大用户来讲是一个不错的选择。微波有机合成实例:http://ng1.17img.cn/bbsfiles/images/2016/09/201609011332_607823_432_3.png反式丁烯二酸与甲醇酯化反应常规加热回流要3小时,产率只有35%。微波加热90秒,产率达68%。http://ng1.17img.cn/bbsfiles/images/2016/09/201609011333_607824_432_3.png羧酸与醚反应常规加热回流要12小时,产率为65%。微波加热35秒,即达相同产率。http://ng1.17img.cn/bbsfiles/images/2016/09/201609011334_607825_432_3.png
多肽合成是一个重复添加氨基酸的过程,固相合成顺序一般从C端(羧基端)向 N端(氨基端)合成。固相合成法,大大的减轻了每步产品提纯的难度。为了防止副反应的发生,参加反应的氨基酸的侧链都是保护的。羧基端是游离的,并且在反应之前必须活化。固相合成方法有两种,即Fmoc和tBoc。由于Fmoc比tBoc存在很多优势,现在大多采用Fmoc法合成。【详情请咨询合肥国肽生物】(1)具体合成由下列几个循环组成:1. 去保护:Fmoc保护的柱子和单体必须用一种碱性溶剂(piperidine)去 除氨基的保护基团。2. 激活和交联:下一个氨基酸的羧基被一种活化剂所活化。活化的单体与游离的氨基反应交联,形成肽键。在此步骤使用大量的超浓度试剂驱使反应完成。循环:这两步反应反复循环直到合成完成。3. 洗脱和脱保护:多肽从柱上洗脱下来,其保护基团被一种脱保护剂(TFA) 洗脱和脱保护。(2)树脂的选择及氨基酸的固定将固相合成与其他技术分开来的最主要的特征是固相载体,能用于多肽合成的固相载体必须满足如下要求:必须包含反应位点(或反应基团),以使肽链连在这些位点上,并在以后除去;必须对合成过程中的物理和化学条件稳定;载体必须允许在不断增长的肽链和试剂之间快速的、不受阻碍的接触;另外,载体必须允许提供足够的连接点,以使每单位体积的载体给出有用产量的肽,并且必须尽量减少被载体束缚的肽链之间的相互作用。用于固相法合成多肽的高分子载体主要有三类:聚苯乙烯-苯二乙烯交联树脂、聚丙烯酰胺、聚乙烯-乙二醇类树脂及衍生物,这些树脂只有导入反应基团,才能直接连上(第一个)氨基酸。根据所导入反应基团的不同,又把这些树脂及树脂衍生物分为氯甲基树脂、羧基树脂、氨基树脂或酰肼型树脂。BOC合成法通常选择氯甲基树脂,如Merrifield树脂;FMOC合成法通常选择羧基树脂如王氏树脂。氨基酸的固定主要是通过保护氨基酸的羧基同树脂的反应基团之间形成的共价键来实现的,形成共价键的方法有多种:氯甲基树脂,通常先制得保护氨基酸的四甲铵盐或钠盐、钾盐、铯盐,然后在适当温度下,直接同树脂反应或在合适的有机溶剂如二氧六环、DMF或DMSO中反应;羧基树脂,则通常加入适当的缩合剂如DCC或羧基二咪唑,使被保护氨基酸与树脂形成共酯以完成氨基酸的固定;氨基树脂或酰肼型树脂,却是加入适当的缩合剂如DCC后,通过保护氨基酸与树脂之间形成的酰胺键来完成氨基酸的固定。(3)氨基、羧基、侧链的保护及脱除要成功合成具有特定的氨基酸顺序的多肽,需要对暂不参与形成酰胺键的氨基和羧基加以保护,同时对氨基酸侧链上的活性基因也要保护,反应完成后再将保护基因除去。同液相合成一样,固相合成中多采用烷氧羰基类型作为α氨基的保护基,因为这样不易发生消旋。最早是用苄氧羰基,由于它需要较强的酸解条件才能脱除,所以后来改为叔丁氧羰基(BOC)保护,用TFA(三氟乙酸)脱保护,但不适用含有色氨酸等对酸不稳定的肽类的合成。chang Meienlofer和Atherton等人采用Carpino报道的Fmoc(9-芴甲氧羰基)作为α氨基保护基,Fmoc基对酸很稳定,但能用哌啶-CH2CL2或哌啶-DMF脱去,近年来,Fmoc合成法得到了广泛的应用。羧基通常用形成酯基的方法进行保护。甲酯和乙酯是逐步合成中保护羧基的常用方法,可通过皂化除去或转变为肼以便用于片断组合;叔丁酯在酸性条件下除去;苄酯常用催化氢化除去。对于合成含有半胱氨酸、组氨酸、精氨酸等带侧链功能基的氨基酸的肽来说,为了避免由于侧链功能团所带来的副反应,一般也需要用适当的保护基将侧链基团暂时保护起来。保护基的选择既要保证侧链基团不参与形成酰胺的反应,又要保证在肽合成过程中不受破坏,同时又要保证在最后肽链裂解时能被除去。如用三苯甲基保护半胱氨酸的S-,用酸或银盐、汞盐除去;组氨酸的咪唑环用2,2,2-三氟-1-苄氧羰基和2,2,2-三氟-1-叔丁氧羰基乙基保护,可通过催化氢化或冷的三氟乙酸脱去。精氨酸用金刚烷氧羰基(Adoc)保护,用冷的三氟乙酸脱去。我们主要提供:多肽合成、定制多肽、同位素标记肽、人工胰岛素、磷酸肽、生物素标记肽、荧光标记肽(Cy3、Cy5、Fitc、AMC等)、目录肽、偶联蛋白(KLH、BSA、OVA等)、化妆品肽、多肽文库构建、抗体服务、糖肽、订书肽、药物肽、RGD环肽等。合肥国肽生物官网:http://www.bankpeptide.com欢迎咨询服务热线:0551-62626599
湖南中医药大学药学院和中南大学化学化工学院的研究人员研究了改进的盐酸舍曲林的合成工艺。他们以α-萘酚为起始原料,经烷基化、甲胺胺化、Pd/C催化氢化、D-(-)扁桃酸拆分,成盐而合成盐酸舍曲林。结果发现:以α-萘酚计,总收率由文献的16.2%提高到31.5%;盐酸舍曲林的结构经红外、差热分析、核磁、质谱确证。该方法工艺简单,适合工业化生产。 蚓激酶提取纯化武汉市畜禽饲料工程技术研究中心武汉工业学院的研究人员建立了高效的蚓激酶纯化工艺,并研究其酶学性质。他们以赤子爱胜蚓(Eiseniafoelide)为原料,经过匀浆抽提、硫酸铵分段盐析、色谱分离、电泳等技术对蚓激酶进行分离纯化及鉴定,并研究pH、温度、金属离子与抑制剂对蚓激酶活性的影响以及蚓激酶的作用机理。结果发现,纯化蚓激酶比活达5100U/mg,经SDS-PAGE电泳分析得2条带,相对分子质量为32000和33500;体外溶栓实验表明,蚓激酶具有直接溶解纤维蛋白和激活纤溶酶原的双重作用。在中性和略偏碱性环境中稳定且活性高;温度适应范围广;不同种类及浓度的金属离子对蚓激酶活性的影响不尽相同;抑制因子及底物特异性研究发现,蚓激酶属于丝氨酸蛋白酶,不属于金属蛋白酶,同时含有二硫键。该分离方法可以得到较纯的蚓激酶。超声提取法优选枳实中橙皮苷提取辽宁医学院药学院和中国医科大学的研究人员研究了优选枳实中的橙皮苷的最佳提取工艺。他们采用L9(34)正交实验,以橙皮苷的含量为标准筛选最佳提取工艺。结果发现:枳实中橙皮苷的最佳提取工艺为采用8倍量的95%甲醇,超声提取3次,每次20min。优选的提取工艺稳定,提取率高。酒石酸拉索昔芬合成中国医药工业研究总院和上海医药工业研究院的研究人员研究了酒石酸拉索昔芬的合成。他们将6-甲氧基-1-四氢萘酮与1-吡咯烷缩合后,经三溴化吡啶鎓溴代,与苯硼酸进行Suzuki偶合、钯炭催化加氢、48%氢溴酸脱甲基后,经D-酒石酸拆分成盐得酒石酸拉索昔芬,以1-吡咯烷计,总收率约为15%。氯维地平合成南京大学化学化工学院的研究人员研究了氯维地平的合成。他们将3-羟基丙腈和双乙烯酮在三乙胺作用下制得乙酰乙酸(2-氰基乙基)酯(2),再与2,3-二氯苯甲醛和3-氨基巴豆酸甲酯经Hantzsch缩合闭环,接着用硫化钠在常温下选择性水解得4-(2,3-二氯苯基)-1,4-二氢-2,6-二甲基-3,5-吡啶二羧酸单甲酯,最后与正丁酸氯甲酯反应即得抗高血压药氯维地平,总收率约为59%。头孢克肟合成浙江普洛得邦制药有限公司和浙江大学化学系的研究人员研究了用7-氨基-3-乙烯基-3-头孢烯-4-羧酸和(Z)-2-(2-氨基噻唑-4-基)-2-甲氧羰基甲氧亚氨基硫代乙酸(S)-2-苯并噻唑酯在三乙胺催化下经酰化、水解反应,“一锅法”制得头孢克肟,收率约为92%。 2票投票
各位: 请教下,尼古丁除了从天然植物提取的方法外,有无人工合成方法?若有,请赐教!谢谢!另外,有关化合物合成方面有没有更专业的网站,介绍下。再次感谢!
1.名称:酯交换法合成甲基丙烯酸高碳烷基酯作 者: 刘福胜 李月刚 穆铁铮 丁文光作者单位: 齐鲁石油化工公司研究院,山东,淄博,255400刊 名: 石油化工 年,卷(期): 2000 29(9) 2.名称:酯交换法合成聚乙二醇单甲基丙烯酸酯 [期刊论文] - 中南民族大学学报(自然科学版) 2007(03)作者:廖国胜.张爱清.雷发泉 3 作者:赵丽燕 论文名称:多元醇双甲基丙烯酸酯的合成及性能研究 [学位论文]硕士 20064,作者:杨斌.赵彩霞.邱宇星.孙东成名称: (甲基)丙烯酸高级醇酯的合成及其应用期刊论文: - 广州化学 2005(04)
1.想请教各位老师,是怎么检测餐具阴离子合成洗涤剂的?以及曲线绘制(详细步骤)2.百度搜到一个说:餐具表面积为A,用100ml水清洗,取25ml水做,最终三氯甲烷定容至25ml。这样是否可以,那这样做曲线应该如何绘制,曲线是取25ml水绘制还是标准所说50ml水绘制?
石油和合成液水分离性测定仪适用标准:GB/T7305 GB/T7605,是测定石油合成液与水分离的能力。液晶屏幕中文显示界面,菜单提示式输入;电脑控温,自动定时,精度高,准确度好;显示年月日及当前时钟等多种参数提示;恒温浴采用小缸体,人性化设计;操作简便,测量准确,外型设计美观;自动搅拌,自动定时,试管搅拌电机大臂自动升降;配有时钟等多种参数提示。仪器特点1.浴缸可随时拆卸,便于清洗和更换2.仪器结构优化,试验过程不损坏试管3.长寿命搅拌电机,机械传动无噪声,稳定可靠4.可同时分离三个样品,提高工作效率5.高清液晶彩屏,全触摸屏操作6.嵌入式linux操作系统7.采用微计算机控制及PID自整定控温技术,控温精度高8.搅拌装置自动升降,减轻了操作人员的劳动强度[font=&]得利特涉及[/font][font=&]多种燃料油分析仪器、绝缘油分析仪器、润滑油分析仪器 (石油和合成液水分离性测定仪、氧化安定性测定仪、密度测定仪、自燃点测定仪、氯含量测定仪、微量残炭测定仪、表观粘度测定仪、机械杂质测定仪),水质分析检测仪器、气体检测仪器,型号多,质量保证,可定制。[/font]
自合成的乙酸异丁酸蔗糖酯的谱图与书中的标准谱图相比,书中的标准谱图在800处有宽吸收带,但是自合成的在该处没有峰,其他吸收峰大多相同。请问着800左右的吸收峰是什么?
[align=center][b]关于阴离子合成洗涤剂的检测方法改进及感想[/b][/align] 随着消毒餐具的发明问世,这种消毒完,包装华丽的餐具很快就广泛被消费者所接受。但是,这种餐具就真的干净吗?随着这种餐具的诞生,阴离子合成洗涤剂的检测也随即问世。阴离子合成洗涤剂又名十二烷基苯磺酸钠,白色或淡黄色粉状或片状固体。 溶于水而成半透明溶液。主要用作阴离子型表面活性剂。今天就和大家分享一下我对对阴离子合成洗涤剂检测方法的见解以及感想。 本次阴离子合成洗涤剂参照《GB/T 5750.4-2006 阴离子合成洗涤剂(第二法)》进行检测,此方法较于第一法省去了繁琐的萃取步骤。标准中要求吸取冲洗餐具表面的水样100 mL进行试验,在样品测定过程中发现,一些商家并为将餐具彻底冲洗干净,导致水样中阴离子合成洗涤剂浓度过高,超出标准曲线的最大点,从而导致检测结果不准确。而并未指出当样品量经前处理后进行上机测定超出线性时的解决办法,为此我们进行了以下实验因此做了以下试验:1、分别吸取100mL水样(平行样)置于2个分液漏斗中,然后分别加入2 mL二氮杂菲、10 mL乙酸铵缓冲溶液、1 mL盐酸羟胺-亚铁溶液和10 mL三氯甲烷(每加入一种试剂均需摇匀),经萃取上机后发现其吸光度大于标准曲线的最大点。2、分别吸取超线性水样25mL置于4个分液漏斗中,其中2个各加入1mL DBS标准溶液作加标回收,4个分液漏斗中的样品并未定容至然后4个分液漏斗中分别加入2 mL二氮杂菲、10 mL乙酸铵缓冲溶液、1 mL盐酸羟胺-亚铁溶液和10mL三氯甲烷(每加入一种试剂均需摇匀),经萃取上机分析后发现其回收率为127%,超出标准要求。3、分别吸取超线性水样25mL置于4个分液漏斗中,其中2个加入1mL DBS标准溶液作加标回收,然后4个分液漏斗中样品用水全部定容至100 mL,再分别加入2mL二氮杂菲、10 mL乙酸铵缓冲溶液、1 mL盐酸羟胺-亚铁溶液和10mL三氯甲烷(每加入一种试剂均需摇匀),经萃取上机分析后发现其回收率为97%,符合标准要求。经过以上几个试验可以得出,当水样浓度过大时,可适当稀释水样,并用纯水定容至100mL,使其吸光度在标准曲线范围内,以得到准确的浓度。 个人感想:在进行阴离子合成洗涤剂检测时,使用的玻璃器具的干净与否直接关系着试验的成败,玻璃器具清洗时,一定不能用洗洁精,洗衣粉等去除污渍的洗涤用品清洗,否则将会严重影响结果。清洗时最好放到酸池里浸泡一段时间,再取出清洗烘干备用。同时注意三氯甲烷有毒,试验时最好在通风橱中进行,以免对身体造成伤害。







 我要推广仪器
我要推广仪器
 下载APP
下载APP