[color=#444444]液相-质谱监测分析中,EI源的[/color][color=#444444]进样瓶定容用二氯甲烷合适吗[/color]
分析氯甲酸乙酯,内标是乙酸异丁酯,固定液为DNP,请问该用什么载体合适?
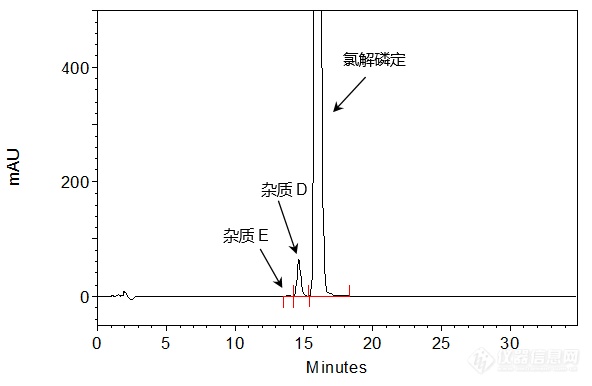
[align=center][b]注射用氯解磷定的有关物质分析[/b][/align]氯解磷定注射液是有机磷中毒解毒药,对急性有机磷杀虫剂抑制的胆碱酯酶活力有不同程度的复活作用,用于解救多种有机磷酸酯类杀虫剂的中毒。[align=center][img=,105,92]http://ng1.17img.cn/bbsfiles/images/2018/07/201807181656260751_676_2222981_3.gif!w105x92.jpg[/img][/align][align=center]氯解磷定[/align][align=center]2-Pyridinealdoxime methochloride[/align][align=center]C7H9ClN2OM.W.: 172.6[/align][align=center][/align]客户提供了注射用氯解磷定样品,希望本实验室依据客户指定色谱条件筛选合适的C[sub]18[/sub]色谱柱,以实现氯解磷定注射液样品的有关物质分析,满足氯解磷定主峰同相邻杂质峰以及各杂质峰间的基线分离要求。首先,尝试使用[b][color=red]中等极性的普适型色谱柱——[/color][color=red]CAPCELL PAK C[sub]18[/sub] MGII[/color][/b],依据客户所提供的色谱条件(流动相含十二烷基硫酸钠和二乙胺)对氯解磷定注射液样品进行分析。如图1,[color=#2E74B5]氯解磷定主峰保留时间为[/color][b][color=#2E74B5]16 min[/color][/b][color=#2E74B5],主峰与峰前杂质[/color][color=#2E74B5]D[/color][color=#2E74B5]之间分离度为[/color][b][color=#2E74B5]2.33[/color][/b][color=#2E74B5],杂质[/color][color=#2E74B5]D[/color][color=#2E74B5]与杂质[/color][color=#2E74B5]E[/color][color=#2E74B5]之间分离度为[/color][b][color=#2E74B5]1.64[/color][/b][color=#2E74B5],均能得到良好分离结果。通过对相对保留时间进行计算,所得结果满足客户[/color][color=#2E74B5]SOP[/color][color=#2E74B5]要求。[/color][color=#2E74B5][/color][align=center][img=,592,386]http://ng1.17img.cn/bbsfiles/images/2018/07/201807181656248670_1537_2222981_3.png!w592x386.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18[/sub] MGII分析所得色谱图[/align][align=center][/align][align=center][/align][align=center][/align][align=center][img=,587,389]http://ng1.17img.cn/bbsfiles/images/2018/07/201807181656253460_3177_2222981_3.png!w587x389.jpg[/img][/align][align=center]图2 CAPCELL PAK C[sub]18[/sub] MGII分析所得色谱图放大图[/align]注:峰上标数字由下至上依次为分离度、理论塔板数与保留时间。[img=,424,282]http://ng1.17img.cn/bbsfiles/images/2018/07/201807181656270060_717_2222981_3.png!w424x282.jpg[/img]就上述实验结果与客户沟通,客户反映希望使主峰的保留时间在20min左右。为满足客户需求,我们在上述实验条件基础上,将柱温由初始条件的40°C降至30°C进行分析,发现主峰保留时间为17 min左右;为进一步增强其保留,我们将色谱柱更换为[b][color=red]极性更高的[/color][color=red]CAPCELL PAK C[sub]18[/sub] AQ[/color][color=red]色谱柱[/color][/b],如图3,[color=#2E74B5]氯解磷定主峰保留明显增强,保留时间约[/color][b][color=#2E74B5]20 min[/color][/b][color=#2E74B5],主峰与峰前杂质[/color][color=#2E74B5]D[/color][color=#2E74B5]之间分离度为[/color][b][color=#2E74B5]2.88[/color][/b][color=#2E74B5],杂质[/color][color=#2E74B5]D[/color][color=#2E74B5]与杂质[/color][color=#2E74B5]E[/color][color=#2E74B5]之间分离度为[/color][b][color=#2E74B5]2.72[/color][/b][color=#2E74B5],能够得到良好的保留与分离结果。[/color][align=center][img=,599,388]http://ng1.17img.cn/bbsfiles/images/2018/07/201807181656255480_6169_2222981_3.png!w599x388.jpg[/img][/align][align=center]图3 CAPCELL PAK C[sub]18[/sub] AQ分析所得色谱图[/align][align=center][img=,597,361]http://ng1.17img.cn/bbsfiles/images/2018/07/201807181656258121_3421_2222981_3.png!w597x361.jpg[/img][/align][align=center]图4 CAPCELL PAK C[sub]18[/sub] AQ分析所得色谱图放大图[/align]注:峰上标数字由下至上依次为分离度、理论塔板数与保留时间。[img=,499,322]http://ng1.17img.cn/bbsfiles/images/2018/07/201807181656269602_541_2222981_3.png!w499x322.jpg[/img]综上实验结果,使用大曹色谱CAPCELL PAK系列色谱柱中的第一选择——中等极性的普适型色谱柱CAPCELL PAK C18 MGII和能在纯水条件下稳定使用的高极性色谱柱CAPCELL PAK C18 AQ进行分析,均能实现注射用氯解磷定的有关物质分析,并能满足氯解磷定主峰同其相邻杂质及各杂质峰间的基线分离要求,客户可根据实际需求进行选择。[align=right]三耀精细化工品销售(中国)有限公司[/align][align=right]技术开发部[/align][align=right]地址:北京经济技术开发区宏达南路5号[/align][align=right]宏达利德工业园1栋418室[/align][align=right]邮编:100176[/align]
[align=center][size=11pt]SEC分析进阶---稳定的分辨率和快速的分析速率[/size][/align][align=center][size=11pt]会议时间[/size][size=11pt]:[/size][size=11pt]2020年2月28日10:00[/size][/align][b][size=10.5pt]内容[/size][size=10.5pt]介绍:[/size][/b][size=10.5pt]尺寸排阻色谱([/size][size=10.5pt]SEC)是广泛使用的聚集体及片段的重要分析手段。在SEC分析的方法开发中,如何获得最优的分离度,保证方法的稳定耐用,以及尽可能提升分析速度,是开发者持续思考的关键问题。本次讲座系统概括了影响以上关键问题的要素并给出了有效建议。[/size][b][size=10.5pt]讲师[/size][size=10.5pt]介绍:[/size][/b][size=10.5pt]米健秋[/size][size=10.5pt]:2004年毕业于北京大学,获得生化分析博士学位。在安捷伦工作12年之久,具有非常丰富的液相色谱,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url],毛细管电泳的应用经历及生物大分子的分析经验。在安捷伦期间作为主要成员参与了2005,2010,2015年版中国药典应用图谱集的工作,主导了多个色谱应用方法开发项目。对于生物制药行业主流应用有着深刻的理解。[/size][size=10.5pt]报名地址[/size][size=10.5pt][font=等线]:[/font][url]https://www.instrument.com.cn/webinar/meeting_10425.html[/url][/size]
[align=center][font=DengXian]分析化学[/font]--[font=DengXian]滴定分析法的滴定液[/font][/align][font=DengXian]滴定液系指已知准确浓度的溶液,它是用来滴定被测物质的。滴定液的浓度用“[/font]XXX[font=DengXian]滴定液([/font]YYYmol/L[font=DengXian])”[/font][font=DengXian]表示。[/font][font=DengXian](一)配制[/font] 1.[font=DengXian]直接法:[/font][font=DengXian]根据所需滴定液的浓度,计算出基准物质的重量。准确称取并溶解后,置于量瓶中稀释至一定的体积。[/font][font=DengXian]如配制滴定液的物质很纯(基准物质),[/font][font=DengXian]且有恒定的分子式,称取时及配制后性质稳定等,可直接配制,根据基准物质的重量和溶液体积,计算溶液的浓度,但在多数情况是不可能的。[/font] 2.[font=DengXian]间接法:[/font][font=DengXian]根据所需滴定液的浓度,计算并称取一定重量试剂,溶解或稀释成一定体积,并进行标定,[/font][font=DengXian]计算滴定液的浓度。[/font][font=DengXian]有些物质因吸湿性强,不稳定,常不能准确称量,只能先将物质配制近似浓度的溶液,再以基准物质标定,以求得准确浓度。[/font][font=DengXian](二)标定[/font][font=DengXian]标定系指用间接法配制好的滴定液,必须由配制人进行滴定度测定。[/font][font=DengXian](三)标定份数[/font][font=DengXian]标定份数系指同一操作者,在同一实验室,用同一测定方法对同一滴定液,在正常和正确的分析操作下进行测定的份数。不得少于[/font]3[font=DengXian]份。[/font][font=DengXian](四)复标[/font][font=DengXian]复标系指滴定液经第一人标定后,必须由第二人进行再标定。其标定份数也不得少于[/font]3[font=DengXian]份。[/font][font=DengXian](五)误差限度[/font] 1. [font=DengXian]标定和复标[/font] [font=DengXian]标定和复标的相对偏差均不得超过[/font]0.1%[font=DengXian]。[/font] 2. [font=DengXian]结果[/font] [font=DengXian]以标定计算所得平均值和复标计算所得平均值为各自测得值,计算二者的相对偏差,不得超过[/font]0.15%[font=DengXian]。否则应重新标定。[/font] 3. [font=DengXian]结果计算[/font] [font=DengXian]如果标定与复标结果满足误差限度的要求,则将二者的算术平均值作为结果。[/font][font=DengXian](六)使用期限[/font][font=DengXian]滴定液必须规定使用期。除特殊情况另有规定外,一般规定为一到三个月,过期必须复标。出现异常情况必须重新标定。[/font][font=DengXian](七)范围[/font][font=DengXian]滴定液浓度的标定值应与名义值相一致,若不一致时,其最大与最小标定值应在名义值的±[/font]5%[font=DengXian]之间。[/font]
我自己开发了一款海水水下叶绿素分析仪器,不知道如何对仪器进行定标,配置叶绿素标准曲线是采用丙酮为基体的叶绿素溶液呢还是培养活体藻类进行稀释测定?如果采用丙酮基体的溶液定标是不是会与实际值差距较大?
求助,分析铝锭中的杂质元素,怎么取样做里面的Fe,Cu,Pb等用什么东西来取,是从铝锭上砸一块下来,还是怎么着?样品取下来,怎么粉碎
冶金氧化铝1000℃灼烧失量(分析标准要求称样量5g),一直以来用“天平—烘箱—天平—马弗炉—天平”的方法分析。此过程较繁琐且结果不稳定,易受环境温湿度、炉温、坩埚位置、转移等因素影响,而且需使用大量昂贵的铂金坩埚。想通过热重分析仪替代旧方法,不知有合适的仪器吗?我认为称样量至少要2g以上(只要分析结果能吻合),如果替代成功,可应用至铝冶炼行业中,市场潜力巨大。chy0999@163.com
如題,CNAS審核不符合項原因分析怎麼回覆比較好?謝謝。不符合項---不確定度評估沒考慮回收率
分析叶绿素a的高速离心机有什么具体参数要求?叶绿素荧光仪能否直接测叶绿素a ?国家环保局认可吗?
液体滴在一定面积的滤纸片或Mylar膜上,用于测定的方法。大家有没有这方面的经验的,能否告知。附件中是聚酯膜微量分析示意图。我的邮箱:xray_mg@sina.com[~76741~]
最近在做一些氧化铝厂物料,铝酸钠溶液的分析实验,有这方面专家请多多讨论.问题:有没有一种不用加热就能分析出铝酸钠溶液中的氧化铝的方法!?
用硫酸,氯酸钠,过氧化氢反应生成二氧化氯消毒剂,采用化学分析法分析残液中的氯酸钠的含量。方法为用硫酸亚铁铵还原氯酸根,再用重铬酸钾滴定未反应完全的硫酸亚铁铵,测定结果含量为12-13%,但按投料比例推算最大含量为17%,是转化率本来就低,还是检测方法的问题。
电镀行业——主要应用在对各种镀液成分的分析。例如氯化物分析,硫酸盐分析,碳酸盐分析,总酸度分析,苛性碱与氧化铝分析,硝酸和HF的分析,硼酸的分析,连续滴定硫酸、铜和铁,连续滴定硝酸、铁和锌,铬分析,锑分析,铁离子和亚铁离子分析,锌分析,钴分析等。食品行业(包括酒、水果、茶和调味料)——主要应用在酸度的分析、水分分析、盐度的分析等等。例如水果中的维生素C分析,茶叶中丹宁酸的分析,淡酒和啤酒的酸度分析,调味料中的山梨酸、柠檬酸的分析、盐度分析等等。铝行业——主要应用在拜尔溶液的分析以及在线控制分析等。例如苛性碱、氧化铝和碳酸盐的分析。造纸和颜料行业—— 主要应用在酸性点和碱性点的分析以及活性酸和活性碱的分析等等。肥料行业——主要是对磷酸及磷酸盐的分析。制药和药物分析行业——主要应用在酸度分析等。例如抗坏血酸分析、阿司匹林分析、水杨酸分析、尿囊素分析、镇静安眠剂的分析等等。漂白行业——主要应用在氯化物、碱度、过氧化氢以及次氯酸盐的分析。聚合物生产工业——主要应用在酸性分析以及对各种聚合物的分析。例如尼龙生产酸度分析、副产品己内酰胺分析以及水分分析;聚亚胺酯工业分析;纤维酸度分析;甲醛分析;酚类分析等等。催化剂行业——主要是采用惰性滴定对催化剂进行相关的研究。例如:酸性点和碱性点的测定、催化剂的热湿分析、添加不同催化剂的反应动力学研究、煤上催化剂的研究、油在颜料上的吸附、溶液热度的测定、Delta H的测定,热容测定、表面积测定等等。清洁剂行业——主要应用在水分析以及主要成分的测定。例如十二烷基硫酸钠(SLS)的测定、非离子清洁剂的测定、水分分析。石油化工行业——主要是在碱度的测定方面。例如石油、沥青、润滑油、变压器油、空调油和矿物或菲矿物油的总碱度的测定、氮荷的测定等等。酸(总酸度)分析——主要应用在弱酸、多元酸和混合酸的连续分析滴定。例如磷酸,磷酸与其它酸的混合物,两种以上混合酸连续滴定分析,硼酸分析等。卤素分析——氯化物、碘化物及氟化物的测定,可应用在海水和电镀相关领域。水分析——水分、化合水及晶格水的测定,可应用在各个需要测定水分的相关领域。金属离子分析——几种金属离子的连续滴定分析、酸和金属混合物的连续滴定分析,可应用在电镀、玻璃制造、金属制造等行业。
请教:做农残分析时,有机氯及菊酯最后用正己烷定容进样分析,有机磷用丙酮或乙酸乙酯定容进样分析。为什么??是与监测器ECD FPD的原因吗?
标定盐酸标准滴定溶液的不确定度分析 作者:吴文英 张春雨 唐惠兰 来源:中华医学研究杂志 在理化分析过程中,一切测量结果都不可避免地具有不确定度。盐酸标准溶液是常用化学定量参比物质,其标定值的准确性直接影响常规分析质量。笔者以GB/T601《滴定分析(容量分析)用标准液的制备》为依据配制并标定盐酸根据JJF1059-1999《测定不确定度评定与表示》分析其测量不确定度。简述由标定过程中得到的不确定度。 1 实验部分 1.1 测定方法[1,2] 准确称量270℃~300℃干燥至恒重的基准碳酸钠(99.95%~100.05%)约0.2g左右,电子分析天平(精度为0.1mg),置于三角瓶中,加入50ml水使之溶解,加指示剂,用盐酸标准液滴定至终点同时作试剂空白实验。 1.2 主要计量仪器与试剂 电了分析天平:AG204;酸式滴定管:50ml A级。 1.3 建立数学模型 C=m (V1-V2)×0.05300 式中 C:盐酸标准滴定溶液的浓度(mol/L);m:基准无水碳酸钠的质量(g);V1:盐酸标准滴定溶液用量(ml);V2:试剂空白实验中盐酸标准滴定溶液用量(ml);0.05300:与1.00ml盐酸标准溶液[C(HCl)=1.000mol/L]相当于以克表示的无水碳酸钠的质量。 1.4 盐酸标准滴定溶液的标定结果 为获得标准溶液重复测量的不确定度分量,对同一标准溶液进行8次独立的标定。测定数据见表1。 表1 盐酸标准滴定溶液的标定结果 略 2 测量不确定度来源 从检测过程和数学模型分析,标定盐酸标准溶液的不确定度主要来源,由四个方面所引起。(1)测量的重复性(A类不确定度);(2)基准无水碳酸钠的纯度;(3)测量使用的电子分析天平及量具;(4)其他相关常数。 3 测量不确定度分析 3.1 A类不确定度的分析 利用表1中的测量结果,按照A类评定测量重复性的标准不确定度。具体计算过程:重复测量的平均值计算式:=1 n∑8 i=1xi=0.09951mol/L 单次测量的标准差按贝塞尔公式计算s(x)为 s(x)=∑8 i=1(xi-)2 n-1=0.0001555mol/L 的标准差s()为 s()=s(x) n=0.000155 8=0.0000548mol/L=5.48×10-5mol/L 由测量重复性引起的相对标准不确定度为U(x):0.0000548/0.09951=0.055%。 3.2 B类不确定度分析 3.2.1 基准碳酸钠的纯度 基准碳酸钠的纯度为1.0000±0.0005,视为矩形分布0.00053=0.00029,则标准不确定度为:由基准碳酸钠的纯度引入的相对不确定度u(p)为:0.029%。 3.2.2 天平称量所引入的标准不确定度 干燥器与天平称量仓内均放置同质硅胶,视为相同湿度,称量时无吸潮。电子天平检定证书标出线性为上0.2mg;可视为矩形分布,则标准不确定度为:因为称量采用的是减量法,故称量的标准不确定度为0.2mg /3=0.12mg:因为称量采用的是减量法,故称量的标准不确定度为:2×0.122=0.17mg,则由称量引入的相对标准不确定度u(m)为:0.17mg/0.2018g=0.084%。 3.2.3 标定体积的不确定度 (1)滴定管的校准:滴定使用50ml酸式滴定管(A级),按照检定规程,其最大允许误差为±0.05ml,相对允许误差为±0.1%,按照矩形分布,则滴定体积的相对标准不确定度u(V)为:u(V)=0.1%/3=0.0577%。(2)环境温度:实验环境在空调条件下,室温近似20℃。温度在20℃左右,标准溶液的温度补正值非常小,对实验结果影响可忽略不计,所以在不确定度分析中不把一温度影响引起的不确定度列入考虑范围。(3)滴定终点的判断:终点时的误差±0.05ml(1滴的体积),两点分布,现由终点分布判断引入的标准不确定度为0.05ml:相对标准不确定度为0.05ml/38.32ml=0.13%标定体积的影响引入相对标准不确定度U(V)为0.0572+0.132=0.142%。 3.2.4 其他常数 基准无水碳酸钠摩尔质量引起的标准不确定度很小,可以忽略。 4 合成标准不确定度 测量重复性、基准无水碳酸钠的纯度、天平称量、标定体积等的不确定度相互独立,故将上述数据合成得盐酸的相对合成标准不确定度U(C)为0.0552+0.0292+0.0842+0.1422=0.176%。 5 扩展不确定度 实验测得盐酸标准溶液浓度为0.09951mol/L,则测量结果的合成标准不确定度U(C)=0.09951mol/L×0.176%=0.000175mol/L。若取包含因子K=2,得测量结果的扩展不确定度U=2U(C)=0.00035mol/L。 6 测量结果的表示 盐酸标准滴定溶液的浓度可表示为:(0.09951±0.00035mol/L,K=2)。 【参考文献】 1 姚正堂,将已峰.奶制品中蛋白质测定的不确定度分析.中华医学研究杂志,2005,5(6):6. 2 国家技术监督局.JJF1059-1999测量不确定度与表示.北京:中国计量出版社,1997,81. 作者单位: 214171 江苏无锡,无锡市惠山区疾病预防控制中心
大家在做重金属ICP分析时候,样品溶液都是先定容再过滤还是先过滤再定容?如何看待这个问题,有标准依据吗?
[color=#444444]原本做一个产品的分析,用的是化学滴定的方法,但是,该方法不是那么准,所以我想改用液相分析,产品是三氟甲基亚磺酸钠,我选的条件是离子柱,流动相是0.1%磷酸水溶液+4g磷酸二氢钾,波长是220nm,检测结果比滴定分析的结果要低4,5个百分点,我想问一下,应该如何选择最佳的色谱条件??[/color]
重熔用铝锭中锌杂质定量分析的化学分析方法是什么?
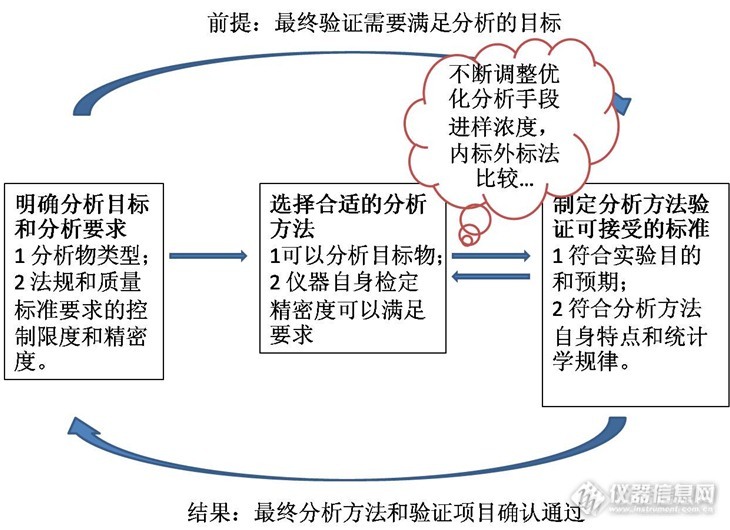
分析方法验证的可接受限度制定 在进行分析方法验证的时候,一般会预先制定的可接受限度,在制定这些验证标准可接受限度的时候需要注意哪些问题呢?我们需要避免哪些“作茧自缚”的不切合实际的限度呢?首先我们从制定分析指标的接受限度联系说起。 1各个验证项目之间的RSD有关联吗? 以液相色谱分析为例,依次要验证的项目是进样精密度、溶液稳定性、重复性和中间精密度。当你设定进样精密度数据要求RSD为5%的时候,重复性验证的标准应该不小于5%。为什么呢?在这里不进行严格的统计证明,通俗的讲的任何一个处理步骤的加入都会引入误差,这些误差是传递和累积的。如: 进样精密度RSD%=仪器检测和进样误差ε; 溶液稳定性RSD%=仪器检测和进样误差ε+检测物溶液稳定误差ε; 重复性误差RSD%=仪器检测和进样误差ε+检测物溶液稳定误差ε+称样误差ε; 中间精密度误差RSD%=仪器检测和进样误差ε+检测物溶液稳定误差ε+称样误差ε+不同操作人员误差ε+不同检测器误差ε+… …… 所以为了符合事实和验证要求,这些验证指标从理论上是逐渐增加的或者至少不变。再次强调一点,我想说的是制定这些指标的应该考虑他们的之间的误差传递情况,考虑验证项目之间的联系,明确验证实验的顺序,不是否定实际实验中真实数据重复性RSD小于进样精密度RSD的情况。统计分析从理论上是考虑的是误差逐渐增大的大多数情况(统计一直考虑大多数),现实情况可能会出现仪器分析带来的误差是正误差,而你称样误差是负误差这种很少见的特殊情况。 2怎样确定进样精密度RSD%? 接上节内容,在明白了验证项目的内在联系和接受标准的误差传递关系后,我们在想:怎样确定进样精密度RSD?其实进样精密度是针对色谱分析方法来说,对所有的分析方法。我们需要知道怎样确定第一个精密度。这第一个精密度简单的来说是量具精密度和仪器检定精密度。 选择合适的量具是保证实验数据准确的前提。天平、移液管、容量瓶等量具是实验的第一步,它们的误差水平是实验误差的开始,一般来说,合适配套的量具使用过程可以控制分析实验(比如常见的滴定实验)的误差在0.3%,高于仪器分析的精密度。 现代药物分析常用的分析手段是仪器分析。仪器检定精密度(仪器性能指标)是各个厂家争相向用户宣传的重要指标,最近的原创大赛很多帖子(在此声明:我不是在做广告)讲了气相色谱,液相色谱和离子色谱的检定过程,检定的指标既是仪器接受和仪器状态评估的重要依据,也是分析方法验证的第一步,在USP和EP中有明确的仪器验证要求,可以根据仪器检定过程确定验证项目的第一个精密度RSD。 仪器检定的精密度是针对系统的,换用不同系统时候这些精密度是需要调整的。以液相色谱为例:紫外的线性和精密度最好(一般可以达到2%),荧光的线性范围较窄,而蒸发光散射检测器(ELSD)精密度达到5%就很吃力,建议设置为7%,我想这也是在欧美药典没有采用ELSD检测器原因之一。除了检测器,同一种仪器采用不同的分析过程也会影响精密度,采用内标法避免了进样过程和信号放大过程的随机误差,一般来说,方法进样精密度比外标法高,严格的气相色谱和LC-MS分析方案一般采用内标法。 3不同数值的RSD有什么不同? 不同数值是什么意思?先看下面两组数据: 数据1514547504851数据2251245247250248251 数据2是在数据1上加200形成的,两组数据的标准误差SD均为2.21,但是数据1的RSD为4.54%,数据2的RSD为0.89%。很多时候一个分析过程的SD是一定的,就是说信号因为系统的干扰变化是一定的。要想数据的变化更多的体现是分析物的变化就必须选择适当的数据输出值,这个数据输出值会影响RSD。调节分析物的绝对含量(称取量、进样浓度体积等)会改变分析的数值。对于液相色谱,一般“调整峰高至满量程的三分之一或者二分之一”,对于滴定分析,20ml的滴定管,一般滴定体积在12-18ml比较合适。如果有的时候输出数值不能调整的合适值怎么办?比如:在用色谱分析有关物质的时候,有关物质色谱峰面积非常小。这个时候你制定可接受的范围就要适当放大,一般来说,低于5个定量限峰高的色谱峰,RSD可以放宽到20%附近。 系统科学的制定分析方法验证的可接受范围[font=宋
我在滴定分析评定不确定度时,涉及滴定管、容量瓶、移液管的不确定度分量时,是考虑检定证书给出的容量示值误差?还是考虑法定允许差啊?整不明白,请高手解答?
请教库仑法分析氯的原理及反应方程式?其滴定池构造如何?
摘要:本文叙述全铝X-射线分析仪分析铝电解质中的Al、F、Na、Ca、Mg含量,进一步计算分子比、CaF2、MgF2、Al2O3、过剩AlF3的方法,以及每个元素及化合物谱线的选择与修正、分析参数的建立、工作曲线的绘制、样品的制备方法等。实践证明:分析结果准确可靠,精密度良好,实现了准确快速测定的目的。一 前言铝槽电解质的分子比是铝电解生产控制的重要参数之一,正确分析电解质的各项指标,直接影响铝电解的工艺控制和经济效益。目前,在国内铝工业生产中铝电解质的分析方法有热滴定法、化学法、结晶光学法和X-射线衍射法,在这些方法中,热滴定法和化学法是基础,但其分析速度慢,分析结果严重滞后;结晶光学法对于有多种添加剂和低分子比的电解质分析时误差太大。X-射线衍射法只有国内少数铝厂采用,其分析的项目较少。本文介绍全铝X-射线分析仪(X荧光+X衍射综合性仪器)分析铝电解质的方法。这是国内从瑞士ARL公司引进的最先进的仪器,经过近一年的实践,证明仪器所分析的数据准确、精密度高、速度快。为青铜峡铝厂三期13万吨200千安预焙电解槽在短时间内达产达标提供了有力的技术支持。使其在4个月内电流效率提高到92%,创造了可观的经济效益。二 实验部分1 实验原理根据邱竹贤、K. Grjotheim等人铝电解质的酸度理论,固态酸性电解质的基体是由冰晶石(Na3AlF6)、亚冰晶石(Na5Al3F14)和Al2O3组成。当加入CaF2时,增加了NaCaAlF6相,液态中增加了CaF2相;加入MgF2时,增加了Na2MgAlF7相,液态中增加了NaMgF3相;加入LiF时,增加了Na2LiAlF6相,液态中增加了Li3AlF6相。因预焙槽工艺中不加LiF,其含量可忽略。根据以上理论,用仪器的荧光部分测定电解质的Al、F、Na、Ca、Mg含量, 再用数学模型计算NaF,AlF3,CaF2,MgF2,Al2O3,过剩AlF3及分子比。2 标样的研制这种标样在实际生产电解槽中直接采取。保证基体相同及每个元素和化合物有足够的梯度。我们在实际生产的640台槽中取样,先用仪器分析其强度,发现单元素有异常的样品,立即大量取样,选取17个单元素有一定梯度的样品,经本厂化验室、郑州轻金属研究所、北京有色金属研究院、包头铝厂、中宁铝厂多家单位化学定值。综合评定,最后选取10个作为标样。3 样品制备为保证分析结果的重复性,从电解槽取样必须严格遵守取样的操作规程。新型全铝分析仪使用慢冷样品,样品中基本上没有非晶质物质存在。各标准样品的冷却条件要和实际取样时尽量保持一致。试样制备过程如下;(1) 粉碎:取电解厂房送来的铝电解质冷却试料块约30g,放入破碎机的试料容器中进行破碎。为了避免破碎时试料粘在容器壁上及压片时易于成型,破碎前滴上1-2滴无水乙醇。经实验在转速1550转/分条件下破碎20秒,可使试料达到300目以上。(2) 压片:将料环放在样托上,称取5克试样粉末倒入料环内,放入压样机,选用30吨压力静压15秒,取出压成的试样片,即可上仪器分析。注意:正常分析样品的取样冷却条件、试样的破碎程度、压样时的压力、静压时间对测量结果均有影响,尽量和标样制备时保持一致。4 选择谱线X-射线荧光是激发原子的最内层K层电子,所以每种元素的特征谱线有好几条,首选Ka谱线,理论Ka谱线与实际生产工艺中元素的谱线并不吻合,必须多做实验加以调整,衍射的谱线也应做调整,无需扣背景,具体谱线见表1。5 确定激发条件对某一种元素,其谱线、晶体、探测器、计数时间、准直器、X-光管电压、电流选择搭配不同,其分析效果也不同。必须做大量实验,总结经验,选择适合生产工艺并能准确反映元素真实含量的分析参数
请问测总氮时,将土壤消解后想用流动分析仪测,是先过滤再定容吗?还是定容后过滤呢
植物纤维中的氯是植物生长的必需元素,我们公司生产的原料基本是植物纤维(纸浆),其氯元素对产品的电气性能会产生不良影响。为了确切知道纸浆中氯的含量我们采取了各种测试方法。1、热水抽提法,将植物纤维(纸浆)在热水(水是经过纯化处置的)抽提一定的时间,然后用各种化学方法进行检测其含量,如硝酸银比浊法、硝酸汞滴定法、离子色谱法。2、将纸浆在更高温条件进行更长时间的抽提,将抽提液进行裂解,再进行离子色谱分析的方法。3、将植物纤维进行燃烧,收集所有烟雾(吸收于水中),然后进行离心,取清的水液进行比浊、滴定及离子色谱分析。上述三类方法中,我们不知道哪一种方法会更客观,也存在一些疑问,请大家一起探讨:1、热水抽提的是水溶性氯物质,植物中应当存在有机形式的氯元素。不管是高温还是沸水都不太可能完全抽提出氯元素,而且氯元素存在形式如果不是离子形式,不管是滴定、比浊还是离子色谱均测试不全面(当然在样品处理时用酸进行反应也可能是可行的。)。2、用燃烧法,再进行酸处理能否对氯元素全部进行离子化反应呢?目前本人还不能完全肯定,但相信反应客观能力上应当比前述的要强些。各位大侠也请发表高论,我们一起讨论讨论吧。
滴定分析容量分析用标准溶液的制备谢谢
石油化工分析仪器系列-- WKL-3000型硫氯分析仪油品中硫氯测定仪SH/T0253产品简介 WKL-3000型硫氯分析仪应用微库仑分析技术,采用氧化法将样品通过裂解炉氧化为可滴定离子,在滴定池中滴定,根据电解滴定过程中所消耗的电量,依据法拉第定律,计算出样品中硫或氯的含量。广泛应用于检测液体、固体或气体样品中的硫氯含量。 仪器具有性能稳定可靠,操作简便,分析精度高,重复性好等特点。技术参数偏压范围:0 ~ 500mv 样品种类:液体、气体和固体 测量范围:S:0.1 ~10000 ng/μl Cl:0.2 ~10000 ng/μl控温范围:室温~1000℃控温精度:±1℃测量精度:样品浓度(ng/μl)进样体积(μl)RSD(%)0.210351.0101010055100052气源要求:普氮和普氧电源要求:AC 220V±22V,50HZ±0.5HZ功 率:3.5KW外形尺寸:主 机:410×350×75(mm) 温 控:530×420×360(mm) 搅拌器:290×270×360(mm) 进样器:350×130×140(mm)
分析有机氯的怪现象分析甲醇中的六六六(甲乙丙丁),db-1701色谱柱,ecd检测器。标准购于国家标准物质中心,单标定性保留时间时发现:甲体保留时间大于丁体约2分钟,与文献出峰顺序不一致,其他均正常,且4种ddd也正常。恳请高手指教!!!!!!
滴定分析法,作为一种简便、快速和应用广泛的定量分析方法,在常量分析中有较高的准确度,滴定分析可算是实验室中最最常用的定量方法啦,很多人都有一个疑问,如果滴定分析结果总是超出了误差范围怎么办?总的来说,对于任何滴定分析,都要首先了解什么样的精度要求才是有意义的并且是必须的,之后如果发现一些结果还是超出了误差范围,你就要从以下几点去找原因:1.待测样品是否在整个样品中具有代表性?换句话说,你应该从取样时就开始寻找可能的错误。“分析结果仅代表实际被分析的样品的结果。”也许在实际测量前,样品可能来自于一个没有混合均匀的容器。亦或在取样后,样品暴露在不同的环境条件下。例如样品在滴定前放置不同的时间段,就会吸收不同量的空气中的二氧化碳。在样品转换器上用敞开式的滴定容器时,就应考虑到这一点。因此我们建议先将滴定容器密闭起来,再在滴定开始之前,用一种特殊装置将其打开(Cover- UpTM),就象Rondo样品转换器上的那种。2. 用多少样品来做分析?对于极少量的样品的分析,天平的性能就至关重要。那么进行一次最小称样量的测试就可以了解天平是否符合要求。3. 如果是滴定仪自身的问题,可从以下几个方面来做检查:a) 馈液管的末端是否有虹吸滴定头,并且工作是否正常?该滴定头是为了防止滴定剂扩散到样品中去。如果失去滴定头,滴定剂就会流入到滴定池中,并和样品反应。但这部分的消耗量是不被计算在内的,因此就能导致比较大的标准偏差。b) 滴定管应检查是否漏气。如果接头没有拧紧或阀的工作不正常,就可能出现漏液。在这种情况下,并不是所有滴定仪馈送的滴定剂都加入到样品中去。由于这种影响不具有重复性,就会导致较大的标准偏差。c) 滴定管中存在有气泡。这通常是由滴定剂中所溶解的气体如CO2、SO2或O2造成的。因此滴定剂在使用前应有个脱气过程,如放置在超声波水浴中。滴定瓶托架作为滴定仪的一个附件可以将滴定瓶提升至与滴定管一样的高度,这就确保在充满滴定管时不会出现负压而造成脱气。卡尔菲休滴定所用试剂由于溶解有SO2,对此极为敏感,因此,在DL31/DL38卡尔菲休滴定仪中,可以适当降低其充液速度。适合滴定分析的化学反应应该具备以下几个条件:(1)反应必须按方程式定量地完成,通常要求在99.9%以上,这是定量计算的基础。(2)反应能够迅速地完成(有时可加热或用催化剂以加速反应)。(3)共存物质不干扰主要反应,或用适当的方法消除其干扰。(4)有比较简便的方法确定计量点(指示滴定终点)。滴定分析法分类1.直接滴定法所谓直接滴定法,是用标准溶液直接滴定被测物质的一种方法。凡是能同时满足上述3个条件的化学反应,都可以采用直接滴定法。直接滴定法是滴定分析法中最常用、最基本的滴定方法。例如用HCl滴定NaOH,用K2Cr2O7滴定Fe2+等。往往有些化学反应不能同时满足滴定分析的三点要求,这时可选用下列几种方法之一进行滴定。2.返滴定法当遇到下列几种情况下,不能用直接滴定法:第一,当试液中被测物质与滴定剂的反应慢,如Al3+与EDTA的反应,被测物质有水解作用时。第二、用滴定剂直接滴定固体试样时,反应不能立即完成。如HCl滴定固体CaCO3。第三,某些反应没有合适的指示剂或被测物质对指示剂有封闭作用时,如在酸性溶液中用AgNO3滴定Cl- 缺乏合适的指示剂。对上述这些问题,通常都采用返滴定法。返滴定法就是先准确地加入一定量过量的标准溶液,使其与试液中的被测物质或固体试样进行反应,待反应完成后,再用另一种标准溶液滴定剩余的标准溶液。例如,对于上述Al3+的滴定,先加入已知过量的EDTA标准溶液,待Al3+与EDTA反应完成后,剩余的EDTA则利用标准Zn2+、Pb2+或Cu2+溶液返滴定;对于固体CaCO3的滴定,先加入已知过量的HCl标准溶液,待反应完成后,可用标准NaOH溶液返滴定剩余的HCl;对于酸性溶液中Cl-的滴定,可先加入已知过量的AgNO3标准溶液使Cl-沉淀完全后,再以三价铁盐作指示剂,用NH4SCN标准溶液返滴定过量的Ag+,出现2+淡红色即为终点。3.置换滴定法对于某些不能直接滴定的物质,也可以使它先与另一种物质起反应,置换出一定量能被滴定的物质来,然后再用适当的滴定剂进行滴定。这种滴定方法称为置换滴定法。例如硫代硫酸钠不能用来直接滴定重铬酸钾和其他强氧化剂,这是因为在酸性溶液中氧化剂可将S2O32-氧化为S4O62-或SO42-等混合物,没有一定的计量关系。但是,硫代硫酸钠却是一种很好的滴定碘的滴定剂。这样一来,如果在酸性重铬酸钾溶液中加入过量的碘化钾,用重铬酸钾置换出一定量的碘,然后用硫代硫酸钠标准溶液直接滴定碘,计量关系便非常好。实际工作中,就是用这种方法以重铬酸钾标定硫代硫酸钠标准溶液浓度的。4.间接滴定法有些物质虽然不能与滴定剂直接进行化学反应,但可以通过别的化学反应间接测定。例如高锰酸钾法测定钙就属于间接滴定法。由于Ca2+在溶液中没有可变价态,所以不能直接用氧化还原法滴定。但若先将Ca2+沉淀为CaC2O4,过滤洗涤后用H2SO4溶解,再用KMnO4标准溶液滴定与Ca2+结合的C2O42-,便可间接测定钙的含量。显然,由于返滴定法、置换滴定法、间接滴定法的应用,大大扩展了滴定分析的应用范围。滴定结果有误,总是预期值的一半或两倍,不知道为什么?这可能有多种原因。结果恰好是预期值的一半或两倍说明这是由于系统误差造成的。首先要做的就是在安装数据中检查为滴定剂所设定的滴定管体积是否与实际相符。滴定剂清单包含所有与滴定剂相关的信息:名义浓度,滴定管体积,所在驱动器以及在滴定度测定后自动储存的当前滴定度值。如果指定的是5mL的滴定管,但实际使用了10mL的滴定管,那么计算结果就只有预期值的一半,反之亦然。 另一种原因可能在于滴定剂的浓度。在结果的计算过程中,名义浓度乘以滴定度才能得到实际浓度,因此错误的名义浓度就可能导致错误的结果。例如:在滴定剂清单中给出的NaOH浓度是0.5 mol/L,而实际上你用的是1.0mol/L的溶液,那么你的结果也就只有预期值的一半了。此外,滴定反应的平衡数z也必须准确,也就是要知道反应的化学计量关系是什么,是不是1:1的反应。错误的平衡数也必将导致结果变成预期值的一半或两倍。终点滴定和等当点滴定有何区别?终点滴定(EP)指传统的滴定步骤:滴定剂持续加入直至反应终止,如用指示剂指定时观察到颜色的变化。对于全自动电位滴定仪来说,持续滴定样品直至达到原先设定的某值,如pH=8.2。等当点是被分析物和试剂的浓度正好相同的那个点。多数情况下,该点完全等同于滴定曲线的回归点,如酸/碱滴定的滴定曲线。曲线的回归点由相应的pH或电位值及滴定剂消耗量(mL)来定义。等当点由浓度已知的滴定剂的消耗量计算得出。通过浓度和滴定剂消耗量能算出已与样品反应的物质的量。全自动电位滴定仪根据滴定曲线应用专用数学评估步骤评估测量点,然后再依据这条评估后的滴定曲线计算出等当点。天平的精度该为多少才能保证获得准确及精
石油化工分析仪器系列-- WKL-3000型硫氯分析仪油品中硫氯测定仪SH/T0253产品简介 WKL-3000型硫氯分析仪应用微库仑分析技术,采用氧化法将样品通过裂解炉氧化为可滴定离子,在滴定池中滴定,根据电解滴定过程中所消耗的电量,依据法拉第定律,计算出样品中硫或氯的含量。广泛应用于检测液体、固体或气体样品中的硫氯含量。 仪器具有性能稳定可靠,操作简便,分析精度高,重复性好等特点。技术参数偏压范围:0 ~ 500mv 样品种类:液体、气体和固体 测量范围:S:0.1 ~10000 ng/μl Cl:0.2 ~10000 ng/μl控温范围:室温~1000℃控温精度:±1℃测量精度:样品浓度(ng/μl)进样体积(μl)RSD(%)0.210351.0101010055100052气源要求:普氮和普氧电源要求:AC 220V±22V,50HZ±0.5HZ功 率:3.5KW外形尺寸:主 机:410×350×75(mm) 温 控:530×420×360(mm) 搅拌器:290×270×360(mm) 进样器:350×130×140(mm)







 我要推广仪器
我要推广仪器
 下载APP
下载APP