苏丹红是应用于油彩、蜡、地板蜡和香皂等化工产品中的一种非生物合成着色剂,不允许在食品中使用。 GB/T19681-2005《食品中苏丹红染料的检测方法/高效液相色谱法》的检验方法标准是比较规范的,有很强的操作性,但在实际运用中,笔者以为还应注意以下几个方面: 溶剂对苏丹红测定结果的影响 1.用乙醚、正己烷作为溶剂配标准溶液时,苏丹红分为苏丹红Ⅰ、Ⅱ、Ⅲ、Ⅳ,其中按国家标准检测方法配制标准溶液母液时,用乙醚溶解后,用正己烷定容,并用正己烷稀释配制标准溶液使用液(系列标准溶液)进行测定。如苏丹红(Ⅰ、Ⅱ、Ⅲ、Ⅳ)混合标准系列溶液不经过氧化铝层析柱处理,直接进样测定时,苏丹红(Ⅰ、Ⅱ)出的峰的峰形很好,但是苏丹红(Ⅲ、Ⅳ)出的峰的峰形较差,甚至低浓度的混合标样中苏丹红(Ⅲ)不出峰,苏丹红(Ⅳ)出的峰不象峰的样子,标准曲线的线性非常差。如果把标准系列溶液同样品一样经过氧化铝层析柱固相萃取处理后蒸干,用丙酮溶解并定容,再进样测定时,苏丹红(Ⅰ、Ⅱ、Ⅲ、Ⅳ)出的峰的峰形非常好,检测结果也理想。 2.用丙酮、乙腈作为溶剂配标准溶液时,分别称取苏丹红(Ⅰ、Ⅱ、Ⅲ、Ⅳ),在丙酮中溶解并定容,用乙腈稀释,再同体积混合并配制混合标准系列溶液进行测定。如苏丹红(Ⅰ、Ⅱ、Ⅲ、Ⅳ)混合标准系列溶液不经过氧化铝层析柱处理,直接进样测定时,苏丹红(Ⅰ、Ⅱ、Ⅲ、Ⅳ)出的峰的峰形非常好,标准曲线的线性也好,检测结果也理想。如果把标准系列同样品一样经过氧化铝层析柱固相萃取处理蒸干后,用丙酮溶解并定容,再进样测定时,苏丹红(Ⅰ、Ⅱ、Ⅲ、Ⅳ)出的峰情况不太好,标准曲线的线性非常差。 氧化铝活化和降活化对苏丹红测定结果的影响 1.氧化铝不活化时,层析用中性氧化铝(100至200目)不活化直接使用做层析柱,后标样和样品通过该层析柱固相萃取处理蒸干后,用丙酮溶解并定容,再进样测定时,色谱图中苏丹红Ⅳ周围杂峰较多,不好判断被测组分的峰。 2.氧化铝活化而不降活化时,取一定量的层析用中性氧化铝(100至200目)放在高30×ф70的称量皿中,在恒温干燥箱中100至105℃活化(烘干)4个小时后,干燥器中冷却至室温做层析柱,后标样和样品通过该层析柱固相萃取处理蒸干后,用丙酮溶解并定容至5mL,经0.45μm有机滤膜过滤后进样测定时,色谱图中苏丹红(Ⅰ、Ⅱ、Ⅲ、Ⅳ)基本上没有峰。 3.氧化铝活化并降活化时,称取一定量的层析用中性氧化铝(100目至200目)放在高30×ф70的称量皿中,在恒温干燥箱中100至105℃活化(烘干)4个小时,于干燥器中冷却至室温后,每100g氧化铝加入2.2mL高纯水,并盖好称量皿的盖子摇匀,放在干燥器中平衡。平衡时间为13至18个小时,摇匀后按标准做法做层析柱,后标样和样品通过该层析柱固相萃取处理蒸干后,用丙酮溶解并定容至5mL,经0.45μm有机滤膜过滤后进样测定时,色谱图中苏丹红(Ⅰ、Ⅱ、Ⅲ、Ⅳ)出的峰形特别好,也没有杂峰,检测结果也理想。 结 论 1.检测过程中,标准物质乙醚溶解用正己烷定容,并用正己烷稀释配制的标准系列溶液不能直接进标样测定,而必须经氧化铝层析柱固相萃取处理蒸干后,用丙酮溶解并定容,再进样测定。 2.检测过程中,标准物质用丙酮溶解并定容,再用乙腈稀释配制的标准系列溶液不能用氧化铝层析柱处理,因为丙酮是解吸液,含丙酮的标准溶液通过层析柱时,吸附、洗涤、解吸过程中,不到解吸步骤,一部分就被测物解吸出去,影响结果,因此直接进样测定就可以。但是样品必须按国标方法处理后进样,固相萃取的样品不能含丙酮。样品处理用的层析柱用氧化铝的活性,用做回收率方式调整最好。 3.层析用氧化铝活化时,应注意活化温度及活化时间,降活化时应注意加水量及平衡时间。样品处理用的氧化铝与标样处理用的氧化铝最好在一个器皿中一次活化,并降活化氧化铝,否则对样品检测结果有影响。

食品中胭脂虫红的测定解决方案胭脂虫红是一种天然色素,主要成分是胭脂虫酸(又称胭脂红酸)。与其他天然色素不同,因其理化性质非常稳定,被视作最安全的天然色素而用作食品、药品、化妆品及纺织品等的着色。猪肉脯在生产加工过程中,常使用色素添加剂,以弥补因光线、空气、加热等原因引起的脱色,并可以提高猪肉脯的感观和对消费者的吸引力,甚至可以通过色素来掩盖制作猪肉脯原材料存在的缺陷或瑕疵。目前国内外胭脂虫红的测定方法主要有反相薄层色谱、光密度扫描分析法、高效液相色谱法、薄层色谱法、分光光度法、毛细管电泳法等。单纯的液相色谱检出较低且干扰较多,通过固相萃取则能弥补这一缺憾。方法优势:迪马科技开发的《食品中胭脂虫红的测定》采用固相萃取-高效液相色谱法测定食品中的胭脂虫红以甲醇为提取液,采用ProElut PLS 固相萃取柱净化样品,通过UPLC检测;本方案前处理步骤简单、净化效果好,重现性好;定量限0.1 mg/kg,远低于《GB 2760-2014 食品安全国家标准 食品添加剂使用标准》对胭脂虫红的限量规定。以下为详细解决方案,敬请参考!食品中胭脂虫红的测定1、适用范围本方案适用于虾条、薯片、蛋卷、果冻和火腿中胭脂虫红的检测。2、提取0.2 g样品(火腿0.5 g),加10 mL 2 mol/L HCl提取液,混匀,75 ℃超声30 min,冷却。加入15 mL 甲醇,振荡,冷却。6000 rpm离心2 min,取上清液。45℃减压蒸至约7 mL,待净化。3、净化——ProElut PLS 60 mg/3 mL(Cat#:68003)a活 化:依次用3 mL甲醇、3 mL水活化;b上 样:加入待净化液,弃去流出液;c淋 洗:加入3 mL 30%甲醇水,抽干;d洗 脱:加入6 mL甲醇,收集流出液;e重新溶解:在45℃水浴下减压蒸至近干,流动相定容至1 mL,供HPLC分析。4、色谱条件色谱柱:Diamonsil C18(2),250 mm × 4.6 mm,5 μm(Cat.#99603)流 速:1.0 mL/min进样量:20 μL柱 温:35 ℃检测器:PDA 483 nm流动相:A:乙腈 B:2%甲酸水溶液 A:B=15:855、添加回收结果食品中胭脂虫红的HPLC检测的添加回收结果样品添加水平(mg/kg)回收率(%)虾条2093.93薯片2087.32蛋卷2085.62果冻2087.56火腿8113.86http://ng1.17img.cn/bbsfiles/images/2016/02/201602011329_584047_1610895_3.jpg胭脂虫红标准(4.0 mg/L)液相色谱图http://ng1.17img.cn/bbsfiles/images/2016/02/201602011329_584048_1610895_3.jpg添加水平为20 mg/kg虾条中胭脂虫红检测的液相色谱图http://ng1.17img.cn/bbsfiles/images/2016/02/201602011329_584049_1610895_3.jpg虾条中胭脂虫红检测(空白样品)的液相色谱图http://ng1.17img.cn/bbsfiles/images/2016/02/201602011329_584050_1610895_3.jpg添加水平为20 mg/kg薯片中胭脂虫红检测的液相色谱图http://ng1.17img.cn/bbsfiles/images/2016/02/201602011329_584051_1610895_3.jpg薯片中胭脂虫红检测(空白样品)的液相色谱图http://ng1.17img.cn/bbsfiles/images/2016/02/201602011330_584052_1610895_3.jpg添加水平为20 mg/kg蛋卷中胭脂虫红检测的液相色谱图http://ng1.17img.cn/bbsfiles/images/2016/02/201602011330_584053_1610895_3.jpg蛋卷中胭脂虫红检测(空白样品)的液相色谱图http://ng1.17img.cn/bbsfiles/images/2016/02/201602011330_584054_1610895_3.jpg添加水平为20 mg/kg果冻中胭脂虫红检测的液相色谱图http://ng1.17img.cn/bbsfiles/images/2016/02/201602011330_584055_1610895_3.jpg果冻中胭脂虫红检测(空白样品)的液相色谱图http://ng1.17img.cn/bbsfiles/images/2016/02/201602011330_584056_1610895_3.jpg添加水平为8 mg/kg火腿中胭脂虫红检测的液相色谱图http://ng1.17img.cn/bbsfiles/images/2016/02/201602011330_584057_1610895_3.jpg火腿中胭脂虫红检测(空白样品)的液相色谱图食品中胭脂红的测定相关产品信息:货号名称规格样品前处理68003ProElut PLS60 g/3 mL, 50/pk24435812管防交叉污染真空SPE萃取装置12位48031,3,6mL柱管通用连接器15/pk4806考克(控制流量)15/pk99011真空/正压两用泵,无油1/pk99013抽滤瓶套装(包括硅橡胶管2米,2L抽滤瓶及橡胶塞
最近,实验室在做染色罐头色素的测定,结果有家企业加入了赤藓红的色素,平时我们最经常做的是诱惑红、胭脂红、苋菜红、柠檬黄、日落黄、亮蓝这几种,发现赤藓红这种色素不能用聚酰胺粉,而我们实验室又没有三正辛胺和阴离子固相萃取柱,问下又没别的方法可以做出来?如果说赤藓红是水溶性的,可不可以直接稀释了上机做呢?这样的回收率应该是不高吧?大家有没有做过的?
1.想简单点购买一台烘箱测定干燥失重,但听说分子内的结晶水是无法在烘箱内烘干的,是吗?2.那么,我就要购买一台水分仪来测定分子内的水,能测定吗?3.如此说来,烘箱和水分仪都要购买了吧?你怎么看!http://simg.instrument.com.cn/bbs/images/brow/em09505.gif
对于分子荧光来说,最大吸收波长移动多少nm才能说发生了红移或蓝移?本人做了不同浓度的蛋白的波长差=15nm的同步荧光光谱,只是浓度不同,并且空白在该波段是没有吸收的,发现浓度变小,最大吸收波长蓝移大概6nm左右,本来没太注意,认为是仪器本身就会有的误差。但是看一文献,是加入其他物质与该蛋白作用后,最大波长蓝移了3nm,然后就得出结论说是酪氨酸残基的微环境改变,其周围的疏水性增强。那对照我做的实验,岂不是不加物质作用的结果还更明显?!本人很困惑,请各位大师指教!
随着标准的更新,GB 5009.35-2016中将诱惑红剔除出了该检测方法,而现行有效的GB 5009.141-2016中采用纸色谱进行分离,分光光度法进行定量,对比两个方法,前处理的原理相近,检测手段发生了改变,因此,有几点疑惑想和各位老师探讨一下:(1)纸色谱与液相色谱相比,有哪些优缺点;(2)液相色谱测定诱惑红有哪些不足之处;(3)您会继续使用液相法测定诱惑红,还是采用新标准中的纸色谱进行分离,分光光度法进行测定。[color=#3f5e09][b]SN/T 1743-2006 食品中诱惑红、酸性红、亮蓝、日落黄的含量检测 高效液相色谱法 该标准目前还是现行有效的[/b][/color]
(1)基本原理: 样品经处理后,试样中的二价钴离子在弱酸性溶液中(pH=5~6)与亚硝基红盐反应生成红色络合物,其颜色的深度与钴含量成正比,用目视比色法进行钴含量的测定,浓度越高,颜色越深,根据光的吸收定律----朗泊-比耳定律,由已知标准溶液浓度对应的吸光值求算出未知浓度: A=K*b*C(2)仪器及试剂:7200型分光光度计 1.0cm比色皿 100 ml容量瓶 5.0 ml、10.0 ml吸量管 0.50ml吸量管250ml烧杯(带表面皿)电炉10%乙酸-乙酸钠缓冲溶液0.2%亚硝基红盐指示剂1:1硝酸 (3)操作步骤:用0.5ml吸量管准确吸取0.2ml料液置于1号250毫升烧杯中,2号烧杯不加料液(做试剂空白),先各加10ml乙酸-乙酸钠缓冲溶液,置于高温电炉上加热煮沸,后置于低温电炉加入亚硝基红盐指示剂5ml,再煮1~3分钟,最后加入1:1硝酸10ml煮1~3分钟,取下冷却,转移至100ml容量瓶中,用蒸馏水定容至100毫升的刻度,摇匀,放置10分钟左右(天热可放置5分钟左右),用1.0 cm比色皿,以蒸馏水做参比,在530nm波长处进行比色,记下空白样的吸光值A空,样的吸光值A样,当钴标浓度为128.8ug/ml时,其吸光值为0.265。一般很稳定(4)计算公式:C(Co2+)=(A样-A空)* 0.1288÷0.265 单位: 克每升C(Co2+)------料液中Co2+的含量,单位:克每升A样-------样的吸光值A空-------空白的吸光值V取样-------取样量(即毫升数)0.1288-----钴标浓度, g/L0.265---钴标浓度为128.8ug/ml时,其吸光值为0.265
今天做合成色素赤藓红的含量,在液相中测定的数据线性,电压值随着浓度的增加反而减小,这是咋回事?柱温30℃,波长设定254
请教一下大神们:小弟最近在做《食品安全国家标准食品中诱惑红的测定》GB5009.141-2016 的扩项工作,无论怎么做都做不好!主要遇到一下几个问题:1.糕点前处理,需不需要去脂肪、蛋白质?(之前去脂肪、蛋白后回收很差);2.前处理净化,之前用过砂芯漏斗效果很差(G3很多残渣,G4会堵),再加滤膜损失很大;3.纸色谱,完全不会。。。。。。(样品点样量太大,形成的色斑巨大;展开剂不懂,标准写的很笼统);敬请各位专家老师指点迷津!!!万分感谢!!!!
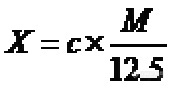
番茄酱中番茄红素快速测定方法 唐玉萍1 范围本非标方法规定了番茄酱中番茄红素快速测定方法。本非标方法适用于在厂检结果对照检验中使用,若本方法测定结果与厂检结果相差超过10mg/100g,则需使用GB/T14215测定。本方法不适合做仲裁检测方法。2 规范性应用文件下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。GB/T 6682 分析实验室用水规格和试验方法GB/T14215 番茄罐头的检验3 原理 番茄红素快速测定仪(LC-01)配置了565nm波长发光二极管(LED),利用漫反射测量原理,可快速测定番茄红素的含量。将样品的可溶性固形物浓度调为12.5%,用番茄红素快速测定仪器测定试样中番茄红素含量,再换算成原浓度样品中番茄红素的含量。4 试剂及材料除非另有说明,所有试剂均为分析纯,水为GB/T6682规定的三级水。5 仪器和设备5.1 番茄红素快速测定仪:LC01,意大利Maselli Misure公司。5.2 折光仪:精度0.5%。5.3 天平:感量0.001g5.4 烧杯:500mL。6 试样的制备和保存6.1 试样的制备将原始样品缩分出1kg,均分成两份,装入洁净容器内,作为试样。密封,并标明标记。6.2 试样的保存将试样于-18℃以下冷冻保存。注: 在制样的操作过程中,必须防止样品受到污染或残留物含量的变化。7 测定步骤7.1 可溶性固形物按GB/T14215规定的方法测定。7.2 制备可溶性固形物含量为12.5%(20℃,Brix)测试样品称取80g均匀的试样,置于烧杯(5.4)中,加适量水稀释,搅拌均匀,使酱体保持适当的悬浮状态,不得有分离现象,调制成可溶性固形物含量至12.5%(20℃,Brix)的样品,待测。7.3 仪器设定开机预热,选择合适的的测试模块(有“热破酱”测试模块“HB”和“冷破酱”测试模块 “CB”之分),进入“HB”或“CB”后,选择”YES”。7.4 样品测定7.4.1 将搅匀的测试样品(7.2)缓缓倒入测试杯中,加入的样品量应超过视窗1cm。查看视窗有没有气泡和污物,气泡必须应清除,必要时用木制的小勺排除气泡。将测试杯放入测试槽,盖好盖板。7.4.2 按“Start Measure”键,开始测试。记录“mg/100g Lyco”栏中的番茄红素值,经原浓度换算后得出原样品中番茄红素含量。7.5 平行试验按以上步骤对同一样品进行平行测定。8 结果计算样品中番茄红素的含量按式(1)计算http://ng1.17img.cn/bbsfiles/images/2011/08/201108021608_308030_1641058_3.jpg…………………………………(1)
[color=#444444]有人用HPLC做辣椒红素的测定吗?我想咨询几个问题,请问辣椒红素的标准品只有国外的吗,大概价位是多少?液相的色谱条件是什么啊?我找的文献中方法都有些不一样,能提供一个具体方法吗?或者提供几个英文的参考文献。麻烦大家了[/color]
各路大神请看过来,情况是这样的,我们有用到人工合成色素诱惑红,标准是“GB 1886.222-2016 食品安全国家标准 食品添加剂 诱惑红”。含量测定我们是用国标中的分光光度比色法,原料到现在应来了好几次,每次含量测定我一般都只能做到75%以上。我看到厂家报告给的数据是85%,国标中的要求也是要求≥85%。我想问问大家有没遇到跟我一样的情况。不是一两次,我还让其他同事也做了,没有一个能把含量做到85%,是分光光度法的问题还是,对照品的问题,又或者是其他问题。对照品是,北京海岸鸿蒙的,浓度1.00mg/mL。
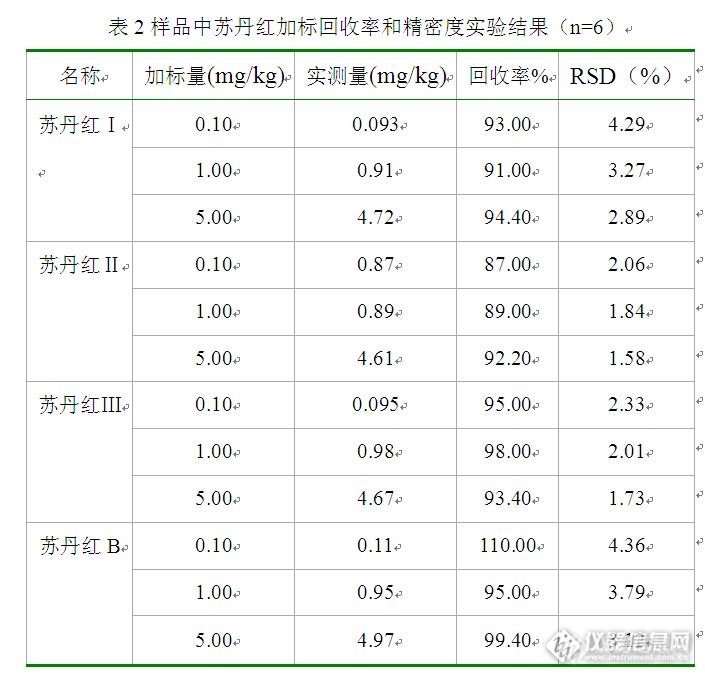
火锅底料中4种苏丹红染料的测定与分析摘要:建立了在线光化学衍生-高效液相色谱法同时测定火锅底料中苏丹红Ⅰ、Ⅱ、Ⅲ和B的方法。样品经丙酮和乙腈的混合溶液(V:V/2:8)提取、C18固相萃取柱净化、高效液相色谱分离后,4种苏丹红染料在紫外光的照射下发生衍生反应,衍生产物进入荧光检测器测定,外标法定量。结果表明,4种苏丹红在0.10~1.0μg/mL范围内线性关系良好,相关系数均大于0.999;低、中、高3个不同加标浓度下, 4种苏丹红的回收率均大于85%,相对标准偏差为1.58%~4.36%;方法的检出限(LOD)为0.30~0.45μg /kg,定量限(LOQ)为1.00~1.50μg /kg。该方法具有重现性好、灵敏度高、结果准确的特点,适用于火锅底料中苏丹红残留的分析检测。目前将光化学衍生应用于苏丹红残留测定的文章鲜见报道。关键词:固相萃取;光化学衍生;高效液相色谱;荧光检测法;苏丹红1 前言 "苏丹红"是一种化学染色剂,并非食品添加剂。它的化学成份中含有一种叫萘的化合物,该物质具有偶氮结构,由于这种化学结构的性质决定了它具有致癌性,对人体的肝肾器官具有明显的毒性作用。苏丹红属于化工染色剂,主要是用于石油、机油和其他的一些工业溶剂中,目的是使其增色,也用于鞋、地板等的增光。由于用苏丹红染色后的食品颜色鲜艳且不易褪色,一些不法食品企业把苏丹红作为色素添加到食品中。 食品中苏丹红染料分析方法有分光光度法、薄层色谱法、高效液相色谱法、液相色谱质谱法和气相色谱质谱法等。国家标准GB/T 19681-2005采用高效液相色谱-紫外法测定食品中的苏丹红,该方法灵敏度较低,在测定时需要切换扫描波长,不利于操作。荧光检测法具有选择性好、灵敏度高,检出限低等优点,在分析科学及其它相关领域的应用越来越多,而苏丹红的荧光响应较低,以荧光检测法测定时需要对其进行衍生,转变为具有较强荧光响应的化学结构。 光化学反应是待测物质在可见光或紫外光的照射下而产生的化学反应,待测物质的分子吸收光子后结构发生转变或者与试剂产生反应,导致化学性质发生变化,与化学衍生反应相比,具有不需要使用额外的衍生试剂,污染较小,选择性好等优点,已被广泛的应用于黄曲霉毒素的测定中,目前将光化学衍生应用于苏丹红残留测定的文章鲜见报道。本文针对火锅底料样品,采用C18固相萃取柱对样品进行浓缩、净化,建立了光化学衍生-高效液相色谱法同时测定火锅底料中苏丹红Ⅰ、Ⅱ、Ⅲ和B的方法,该方法具有重现性好、灵敏度高、结果准确的特点。2 材料与方法2.1 仪器及设备 Waters e2695高效液相色谱仪(主要包括2475荧光检测器,柱温箱,自动进样器等);光化学衍生器(由紫外灯、聚四氟乙烯反应管组成);超生波清洗;离心机;旋转蒸发器;氮气吹干仪;C18 (500mg/6mL)固相萃取柱。2.2 试剂 乙腈、甲醇和丙酮,色谱纯;无水硫酸钠,分析纯;实验用水为Millpore超纯水; 标准物质:苏丹红Ⅰ(纯度≥99.0%),苏丹红Ⅱ(纯度≥99.0%),苏丹红Ⅲ(纯度≥96.0%),苏丹红B(纯度≥90.0%)。2.3 分析测定条件 色谱柱: C18 (250mm×4.6mm ,5μm);流动相A为乙腈,B为甲醇,梯度洗脱:0~11min,100% A;11~12min,100% A~0%A,0%B~100%B;12~25min,100% B;流速:0.8mL/min;进样体积:20μL;柱温:30℃;荧光检测器:激发波长310nm,发射波长348nm。2.4 样品前处理2.4.1 样品的提取 称取2.0g试样于50.0mL离心管中,加入5.0g无水Na2SO4, 20.0mL丙酮-乙腈(V:V/2:8)溶液,超声提取20min,以5000r/min离心5min,上清液转移至250mL鸡心瓶中,残留物用20.0mL丙酮-乙腈溶液重复提取一次,合并上清液,并在40℃水浴中减压旋转蒸发浓缩至约1mL,待净化。2.4.2 固相萃取柱富集净化 C18固相萃取柱依次用5mL乙腈、5mL水进行活化和平衡,然后将上述待净化液上样过柱,用1mL丙酮-乙腈溶液润洗鸡心瓶,并将润洗液转移至固相萃取柱中,用5mL水淋洗,再用5mL丙酮-乙腈溶液进行洗脱,收集全部洗脱液,氮气吹干,用丙酮-乙腈溶液重新定容至1mL,经0.45μm滤膜过滤后进样分析。2.5 标准溶液的配制 分别准确称取一定质量的苏丹红Ⅰ、Ⅱ、Ⅲ和B,加入少量丙酮超声溶解后转入100mL容量瓶中,用乙腈稀释成浓度为0.10mg/mL的标准储备液,放置于4℃冰箱中避光保存。临用前,分别准确吸取一定体积的0.10mg/mL的标准储备液,用乙腈逐级稀释到相应浓度的混合标准溶液。3.结果与讨论3.1 扫描波长的选择 采用Waters 2475荧光检测器自带的3D扫描功能分别在波长200-340nm、330-600nm范围内扫描4种苏丹红光化学衍生产物的激发波长和发射波长,扫描结果见图1。从图中看出,苏丹红Ⅰ、Ⅱ、Ⅲ和B经光化学衍生后其衍生产物的最佳激发波长分别为312nm,312nm,305nm,305nm;最佳发射波长均为348nm,因此本文选择激发波长310nm发射波长348nm作为扫描波长对4种苏丹红进行同时测定。http://ng1.17img.cn/bbsfiles/images/2016/09/201609171416_610070_1669358_3.jpg3.2检测方法的选择 分别采用荧光检测器和柱后光化学衍生-荧光检测器对相同浓度的4种苏丹红混合标准溶液进行测定,测定结果如图2。从图中可以看出,采用荧光检测器测定时4种苏丹红仅表现出微弱的荧光响应,不利于痕量组分含量的测定;经光化学衍生后4种苏丹红的荧光响应大为增加,这是由于苏丹红是一类萘酚偶氮结构型染料,其分子结构中的偶氮基团具有光化学活性,在光照作用下能够发生顺式和反式结构的互变,甚至能够与溶剂发生光化学反应,从而导致偶氮化合物的性质发生显著的变化,生成具有较强荧光响应的物质。http://ng1.17img.cn/bbsfiles/images/2016/09/201609171417_610071_1669358_3.jpg3.3 流动相的选择 分别以乙腈、甲醇、乙腈-水(V:V/98:2)、甲醇-水(V:V/98:2)、乙腈-甲醇(V:V/98:2)为流动相等度洗脱,光化学衍生后测定,考察4种苏丹红的出峰及分离情况,结果如图3。以乙腈为流动相时,4种苏丹红均能产生较强的荧光响应,且4种苏丹红具有很好的分离度;以甲醇为流动相时,苏丹红Ⅰ和苏丹红Ⅱ仅有微弱的荧光响应,响应值较低,难以满足苏丹红残留的检测要求,苏丹红Ⅲ和苏丹红B具有较强的荧光响应,与乙腈为流动相相比其响应值更高;以乙腈-水(V:V/98:2)、甲醇-水(V:V/98:2)、乙腈-甲醇(V:V/98:2)为流动相时,苏丹红Ⅰ和苏丹红Ⅱ的荧光响应均较弱,与未发生光化学衍生反应测定的响应值相近。上述实验表明苏丹红的光化学衍生反应及反应产物的荧光性质与参加反应的溶剂相关,苏丹红Ⅰ和苏丹红Ⅱ在纯乙腈条件下才能发生光化学衍生反应,当流动相中含有微量的水和甲醇时都会影响其光化学反应;苏丹红Ⅲ和苏丹红B与甲醇和乙腈均能发生光化学反应,微量的水不影响这两者的光化学反应,而且在甲醇流动相中两者的光化学衍生产物的荧光响应值更高。主要原因是随着苏丹红Ⅲ和苏丹红B分子结构中共轭体系的增大,分子极性减弱,从而在极性较弱的甲醇中表现出更强的荧光强度。因此分别选择乙腈和甲醇为光化学反应的溶剂,采用梯度洗脱的方法对4种苏丹红进行分离和测定。http://ng1.17img.cn/bbsfiles/images/2016/09/201609171424_610072_1669358_3.jpg3.4 样品前处理条件的选择 现有的国家标准测定苏丹红时通常采用乙腈直接提取或者氧化铝层析柱净化的方法进行样品前处理,而前者容易受到样品基质的影响,提取后存在大量的干扰物质,影响目标物质的定性和定量;后者处理方法较为繁琐,氧化铝的活性对方法回收率的影响很大。图4为空白样品和加标样品采用C18固相萃取柱净化后测定的色谱图。实验表明,C18固相萃取柱对4种苏丹红具有很好的保留作用,以水为淋洗液可以一次性去除样品中大部分水溶性干扰物质,减少杂质对苏丹红检测结果的影响,同时采用C18固相萃取柱进行样品前处理,对待测组分还具有浓缩和富集的作用,有利于降低待测组分的检出限。http://ng1.17img.cn/bbsfiles/images/2016/09/201609171427_610073_1669358_3.jpg3.5标准曲线、线性范围及检出限 分别准确吸取适量体积的苏丹红标准储备液,用乙腈逐级稀释成一定浓度的混合标准溶液,按上述优化的实验条件进行测定,以峰面积(y,μV·s)对
各位,大家用原子吸收测铅的时候用到的玻璃仪器都是怎么干燥的呀?不能用烘箱烘干吗?以前听别人说不能用烘箱,夏天还比较好自然干燥,现在想要自然干燥是在是太难了不能用烘箱的原因是什么呀?烘箱会带入杂质铅吗?
一、案例2005年2月18日,英国最大的食品制造商第一食品公司生产的沙司中发现了被欧盟禁用的“苏丹红一号’’色素。而这些沙司又卖给了大量食品厂商和超市卖场。从此,苏丹红成为全世界食品安全问题的代名词。苏丹红事件已过去几年了,但随后与苏丹红相关联的食品安全事件还时有发生,如红心鸭蛋等,有效的预防措施是随时进行检查,将可能受到苏丹红污染的食品杜绝在人们的消费前,才能保证食品消费的安全。二、选用的国家标准GB/T 1968l一2005食品中苏丹红染料的检测方法——高效液相色谱法。三、测定方法1.样品处理(1)粉状样品(如辣椒粉等) 准确称取样品1.000~5.000g于锥形瓶中,加入10~30mL正己烷,超声过滤5min,再用10mL正己烷洗涤残渣数次,至洗出液无色,合并正己烷液,用旋转蒸发仪浓缩至5mL以下,慢慢加入氧化铝色谱柱中(氧化铝色谱柱的制备方法:在色谱柱管底部塞入一薄层脱脂棉,干法装入处理过的氧化铝3cm高,轻敲实后加一薄层脱脂棉,用10mL正己烷预淋洗,洗净柱中杂质后,备用),为保证层析效果,在柱中保持正己烷液面为2ram左右时上样,在层析过程中保持柱的湿润,用正己烷少量多次淋洗浓缩瓶,一并注入色谱柱;控制氧化铝表层吸附的色素带宽宜小于O.5cm,待样液完全流出后,视样品中含油类杂质的多少用10~30ml。正己烷洗柱,直至流出液无色,弃去全部正己烷淋洗液,用含5%丙酮的正己烷液60mL洗脱,收集、浓缩后,用丙酮转移并定容至5mL,经0.45μm有机滤膜过滤后待测。(2)油状样品(如红辣椒油、火锅料、奶油等)称取0.500~2.000g样品于小烧杯中,加入适量正己烷溶解(1~10mL),难溶解的样品可于正己烷中加温溶解,其余同上操作。(3)含水量较多的样品(如辣椒酱、番茄沙司等) 称取10.00~20.00g样品于离心管中,加10~20mL水将其分散成糊状,含增稠剂的样品多加水,加入30mL正己烷:丙酮(3:1),匀浆5min,3000r/min离心10min,吸出正己烷层,下层再分别用20mL正己烷匀浆两次,离心后合并3次正己烷,加入5g无水硫酸钠脱水,过滤后于旋转蒸发仪上蒸干并保持5min,用5ml,正己烷溶解残渣后,其余同上操作。(4)肉制品(如香肠等) 称取粉碎样品10.00~20.00g于锥形瓶中,加入60mL正己烷充分匀浆5min,滤出清液,再分别用20mL正己烷匀浆两次,过滤后合并3次滤液,加入5g无水硫酸钠脱水,过滤后于旋转蒸发仪上蒸至5ml。以下,其余同上操作。2.样品测定(1)色谱条件①色谱柱:ZorbaxSB—C18 3.5μm,4.6mm×150mm(或相当型号的色谱柱)。②流动相:溶剂A(0.1%甲酸的水溶液:乙腈===85:15);溶剂B(O.1%甲酸的乙腈溶液:丙酮一80:20)。③流速:1mL/min。④柱温:30℃。⑤检测波长:苏丹红I 478nm;苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ520nm,于苏丹红I出峰后切换。⑥进样量:10μL。(2)标准曲线的制作 吸取标准储备液O、0.1ml、O.2ml、O.4mL、O.8mL、1.6mL,用正己烷定容至25mL,此标准系列使用液浓度为O、0.16μg/mL、O.32μg/mL、0.64μg/ml、1.28μg/mL、2.56μg/mL,取10μL按色谱条件进样测定,并绘制标准曲线。(3)样品测定 依色谱要求条件,进样操作,测得结果,代入公式计算。3.结果计算R=C×V/m式中 R——样品中苏丹红的含量,mg/kg;C——由标准曲线得出的样液中苏丹红的浓度,μg/mL;V——样液定容体积,mL;m——样品质量,g。4.试剂①乙腈:色谱纯。②丙酮:色谱纯、分析纯。③甲酸:分析纯。④乙醚:分析纯。⑤正己烷:分析纯。⑥无水硫酸钠:分析纯。⑦色谱柱管:lcm(内径)×5cm(高)的注射器(管)。⑧色谱用氧化铝(中性100~200目):105℃干燥2h,于干燥器中冷至室温,每100g中加入2mL水降活,混匀后密封,放置12h后使用。⑨5%丙酮的正己烷溶液:吸取50mL丙酮用正己烷定容至1L。⑩标准储备液:分别称取纯度≥95%的苏丹红I、苏丹红Ⅱ、苏丹红Ⅲ及苏丹红Ⅳ各10.Omg(按实际含量折算),用乙醚溶解后用正己烷定容至250mL。5.仪器①高效液相色谱仪(配有紫外可见光检测器)。②分析天平:感量0.1mg。③旋转蒸发仪。④均质机。⑤离心机。⑥O.45μm有机滤膜。
有个测定润滑油中酚含量的行业标准,方法是这样的:用水将油中的苯酚萃取出来后,加入NaOH溶液,用287nm和350nm的吸光度之差作曲线和测定。我知道287nm是苯酚红移后的最大吸收谱线之一,但为什么不用最大吸收的235nm?350nm是如何得到的?请教专家指点!
同一溶剂下,带有不同取代基的化合物的紫外吸收峰发生红移的原因有哪些?
请教各位,有个测试项目测水分,我们使用水分测定仪,代替烘箱和天平,申报实验室认可行不行呢?
如题 如何测定汤圆中胭脂红啊? 前处理怎么操作啊
各位大哥,有做过番茄中番茄红素测定的,请指点一二?
食品中红糟的测定方法急需!谢谢!
征集食品中苏丹红各种测定方法,另外把测定中的问题疑问大家都来谈谈我从北分厂技术应用部得到了一个比国标简单的方法.大家也把自己的测定方法拿来谈谈,多说说测定中的问题.http://www.instrument.com.cn/download/shtml/030784.shtml希望搂住赶快结帖,否则给与惩罚
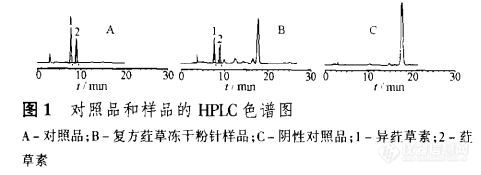
【作者】 兰燕宇; 王爱民; 何迅; 李勇军; 刘丽娜; 王永林; 【Author】 LAN Yan-yu, WANG Ai-min, HE Xun, LI Yong-jun, LIU Li-na, WANG Yong-lin(School of Pharmacy, Guiyang Medical College, Guiyang 550004, China) 【机构】 贵阳医学院药学院; 贵阳医学院药学院 贵州 贵阳 550004; 贵州 贵阳 550004; 贵州 贵阳 550004; 【摘要】 目的建立注射用复方荭草冻干粉针中异荭草素、荭草素含量的RP—HPLC测定方法。方法色谱柱:Diamonsil ODS(4.6 mm×250 mm,5 μm);流动相:乙腈-0.1%磷酸溶液(18:82),流速:1 mI·min-1,柱温:40℃;检测波长350 nm。结果被测定峰与其他组分峰可达到基线分离,异荭草素、荭草素线性范围分别为0.049 6~0.794 0μg(r=0.999 9),0.031 6~0.506 0 μg(r= 0.999 9),回收率分别为98.4%,99.9%,RSD分别为2.2%,1.3%(n=9)。结论该法简便、准确、重现性好,适用于该制剂的质量控制。 【关键词】 反相高效液相色谱法; 复方荭草冻干粉针; 异荭草素; 荭草素; http://ng1.17img.cn/bbsfiles/images/2012/09/201209022122_388015_1838299_3.jpg
五氯酚藏红T分光光度法的测定,直接买现成的五氯酚钠标液可以吗?所有步骤按照国标做的,可是就是不显色的原因可能有哪些呀?
[color=#444444]紫外可见吸收光谱中,比如Au失电子变为Au3+,吸收峰是红移还是蓝移啊???[/color]
中国药典上关于对水分测定烘干法的表述是这样的:“取2-5g,……精密称定”,指的是否可用千分之一天平来进行水分的测定实验?
求助胭脂红的测定,2760中胭脂红及铝色淀结果以胭脂红计,目前有没有检测铝色淀的标准啊,5009.35只能检测胭脂红。但判定时是以两个物质之和来定量的。求高手们指点
我欲选购如下几种沥青性质分析仪器: 薄膜烘箱分析仪,要求比老式仪器有更好的性能。 自动软化点测定仪。 延长度测定仪。 欢迎各厂家提供详细信息,包括仪器的性能,价格,适用标准,还有哪些厂在使用等等。 原来使用的仪器有些老旧,有的性能无法达到标准要求需更换。 急需这方面的信息!!!请尽快提供。
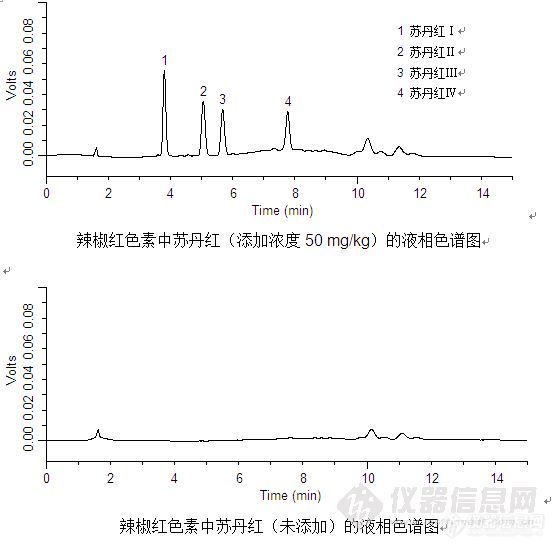
辣椒红色素中苏丹红的测定1 序言苏丹红是一种人工合成的染料,为亲脂性偶氮化合物,主要包括Ⅰ、Ⅱ、Ⅲ和Ⅳ四种类型。常作为一种工业染料,在食品中非天然存在,如果食品中的苏丹红含量较高,达上千毫克,则苏丹红诱发动物肿瘤的机会就会上百倍增加,特别是由于苏丹红有些代谢产物是人类可能的致癌物。早在2003年5月,法国发现从印度进口的红辣椒产品中含有苏丹红Ⅰ号。随后,在欧盟成员国加强检测后,又在包括咖喱粉在内的一系列进口辣椒产品中发现了苏丹红Ⅱ、Ⅲ和Ⅳ号。2005年2月,英国食品标准局在英国“第一食品公司”制造的伍斯特郡辣酱油使用的辣椒粉中查处了苏丹红Ⅰ号。并于2月下旬,向全球发出十五安全警告。随后,我国也加强了苏丹红Ⅰ~Ⅳ号的检测工作,发布了“食品中苏丹红染料的检测方法-高效液相色谱法”国家标准。2 样品准备/提取(1) 称取0.1 g~0.2 g样品于离心管中,加入10 mL乙腈;(2) 涡旋混合5 min,超声提取5 min,6000 rpm下离心3 min,收集上清液;(3) 残渣再用10 mL乙腈提取,每次涡旋混合5 min,超声提取5 min ,6000 rpm下离心3 min;合并两次提取液;(4) 在40 ℃下用减压蒸馏将提取液蒸干,然后用5 mL正己烷溶解,待净化。3 SPE柱净化——ProElut Silica 1 g/6 mL(Cat.#:63006) (1)活 化: 10 mL正己烷,流出液弃去; (2)上 样: 将待净化液加入小柱,流出液弃去; (3)洗 脱: 15 mL乙醚:乙腈=1:9洗脱,收集流出液; (4)重新溶解: 在40 ℃下用减压蒸馏将收集的流出液蒸干,然后用乙腈定容至2 mL后供HPLC分析。 4 分析条件 色谱柱: Diamonsil C8(2) 150×4.6 mm,5 μm(Cat.#:99650) 流 速: 1.0 mL/min [align
[color=#0000ff][size=4] 我想就药典中新增的安宫牛黄丸中胆红素含量测定方法问题向大家请教:为什么我们在试验中胆红素含量会很低呢?(原料牛黄中胆红素含量不低呀)操作中应该注意些什么呢?(避光,低温。) 急呀![/size][/color]







 我要推广仪器
我要推广仪器
 下载APP
下载APP