甘露醇检测时先出哪个峰?在检测甘露醇时是先出甘露醇的峰还是先出山梨醇的峰?
流动相为纯甲醇,样品为纯甲醇时,没有进样峰,在2.1min除了一个峰,这个峰会是怎样引起的呢?甲醇换个几瓶,仪器也换了,结果都一样,应该和甲醇没关系。走空针时,很干净,应该和柱子也没关系了。样品为纯乙腈时,有进样峰,还是有这个峰。求高人指点呀!!!
做白酒时,甲醇、乙醇出峰分不开,分别打色谱级甲醇、乙醇,它们的出峰时间基本相同,打标样时只出一个峰,要怎么调整才能做出两个峰呢?我用的是福立9790,DB-FFAP毛细管柱,柱温170℃(恒温),检测器190℃,注样器190℃。有人说可以用程序升温,但我的标样里含有水,打进去会不会损坏色谱柱呢?要是不会损坏,那我程序升温怎么设置?
毛细管电泳用甲醇做溶剂,出的甲醇系统峰,可以认为是中性物质出峰时间么,若样品出峰时间晚于系统峰出峰时间,可以认为样品是带负电的么?
新手求助,在进行杂醇油出峰时间确定的时候发现异戊醇的峰是3个峰连一起的这是正常的情况还是有杂质干扰。
现在做白酒中甲醇含量的测定,加标品后(0.02%体积分数)甲醇拖尾导致峰型很难看,以前做其他物质的时候有甲醇峰出现的时候也是如此,其他物质的峰型还不错。请高手给指点一下。仪器参数:安捷伦6890,进样口250,检测器FID250,程序升温:35度1min,3.5℃/min升到120。柱子是 VF-Wax 60*0.25*0.25。甲醇峰在10.3min左右。
芳樟醇 CAS:根据CAS确定的物质一般是指一种纯物质吗?本人用GC-MS分析芳樟醇的标准溶液,采用不同的程序升温,结果不同。以15℃/min分析时,谱图上就一个峰。但以4℃/min分析时,谱图上有四个峰,这是什么个状况啊?四个峰的碎片离子基本一样,就是丰度不同。
流动相是甲醇和水 比例为45:55待测样品用甲醇溶解每次都有一个不明的峰 在同一位置 怀疑是甲醇1、是甲醇吗2、能把它当背景去掉吗 因为有其它峰和她有点重叠
我用的强极性毛细管柱,苯峰在甲醇的拖尾处,乙醇的峰与甲醇又很近,如果乙醇含量高的话,又会积分成苯的峰。我应该用什么极性的柱子分离甲醇(空白溶剂)中乙醇与苯的峰呀,非极性柱子吗?
[url=https://insevent.instrument.com.cn/t/Mp]gc[/url] 甲醇峰和乙醇峰之间有个出峰信号,实验了多种溶剂都不符合,想求助会是什么溶剂或者物质?
流动相是70%甲醇和30%水,进纯甲醇和水在同一时间出峰,但是流动相换成是70%乙腈和30%水,进纯甲醇时就会出来两个峰,这是怎么回事呢?麻烦大家能帮帮我呀,如果是溶剂峰的话为什么用乙腈是两个峰呢?我是色谱新手,多谢大家了

[img]http://ng1.17img.cn/bbsfiles/images/2009/06/200906301620_157855_1768156_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/06/200906301620_157856_1768156_3.jpg[/img]如何提高 白酒甲醇测定中 甲醇峰踏板数?图和系统评价在上面2#峰(保留时间3.332)为甲醇峰;4#峰(保留时间4.948)为内标正丙醇峰.试过很多条件,甲醇峰的塔板数一直不理想。请问各位有什么好的办法提高甲醇的塔板数?仪器为鲁南瑞虹6890,载气流速旋钮无效,但是可以调节柱头压中科安泰 毛细管柱、分流进样。
测白酒里的异戊醇,出峰的时候后边一直有一个小峰,加标回收后小峰面积基本不变,但是加标后异戊醇峰甚至会变得更小,加标回收率也一直不稳定,有没有大佬帮帮我,太难了??。
顶空GC-FID db-wax 柱子分析水中甲醇,以前的相同浓度的标样的峰面积都在18左右,前几天色谱出了故障,色谱柱拆到另一台色谱柱上用过,仪器修好之后,又重新换上原来色谱柱,甲醇的峰面积变为了2左右,将甲醇浓度增加10倍,峰面积仍为2左右,保留时间没有变化,似乎已没有线性关系。而这根色谱柱分析苯系物均没有问题。难道色谱柱会有选择性的坏?求各位指点,不胜感激。
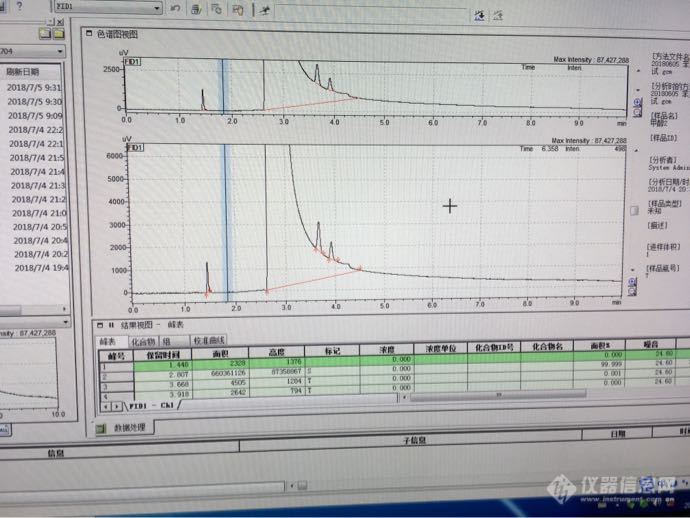
我们用的是岛津GC2030,买了HPLC级甲醇,现在做验收,走出来的峰如图,甲醇峰上有两个小峰,我做的项目是苯系物,不知道拿来配标液会不会有影响,请问验收合格的依据是啥?[img=,690,517]http://ng1.17img.cn/bbsfiles/images/2018/07/201807050938052668_5569_3019776_3.png[/img]
峰纯度是如何计算的?因为样品峰型(样品峰型图及其光谱图请见[url]http://bbs.instrument.com.cn/shtml/20100728/2689015/[/url]图7、图8)不是很好,但是以顶峰(样品色谱图的4.806分钟峰)时紫外可见吸收光谱(见同一贴图8)建库,然后检索峰起始至峰结尾时不同保留时间时UV-Vis光谱图,发现重合都很好,但是报告模板中计算结果则显示峰纯度不高,何解?
同为极性柱子,为什么乙醇叔丁醇在PEG20M的柱子上乙醇出峰在前,FFAP的柱子顺序相反呢?
气相色谱岛津2014分析甲醇和乙醇含量,毛细管柱,柱温55,进样口120,检测器160,已经做过一年多了都很正常,中间一个多月没有用柱子,再用就有很多杂峰,老化处理,换了石英棉后,杂峰不见了,可是甲醇乙醇峰分不开了,请教高手
用FID测乙醇中的甲醇,乙醇出峰吗?

用极性柱分离极性物质时,应该是极性小的物质先出峰,但是甲醇和乙醇相比,应该是乙醇的极性小,怎么是甲醇先出峰呢?
各位老师,按理说甲醇比乙醇极性大。为啥在极性柱上,确实甲醇先出峰,乙醇后出峰ne
流动相使用95%甲醇+5%水,不进空白试剂时一级质谱TIC图中没有出现峰,当空白溶剂使用甲醇(质谱级)或乙腈(色谱级)并进质谱时,两个不同溶剂居然在TIC图中的相同位置出峰,并且峰对应的质谱图主峰相同,从色谱柱后面接一小瓶流动相作为空白试剂进样没有出峰,柱子是刚换的新柱子,不可能出现污染等问题,甲醇和乙腈不是不应该出峰的吗?请各位大佬帮忙分析一下,感谢
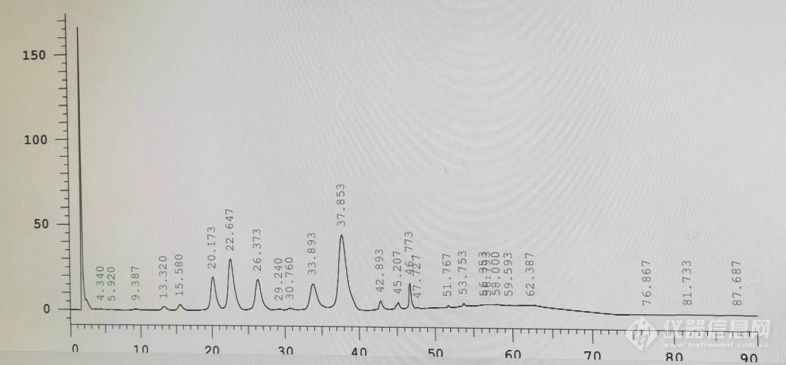
各位同行,请教下,我按照中国药典做紫杉醇有关物质时,进样浓度500μg/ml,主峰一般在37.8min左右出峰,其中一次进样,20-30min出了好几个峰,主峰响应值也比平时要低,峰面积加起来和正常进样(只有37.8min出峰时)那个峰面积差不多的,这是从来没有过的,奇怪的是,隔天同样样品再次进样这几个峰就没了,原因有没可能是紫杉醇样品分段出峰了?是不是柱子问题,还是其他原因?有没大佬帮忙分析下,不胜感激。[img=,690,320]https://ng1.17img.cn/bbsfiles/images/2022/08/202208091546131480_9156_5567255_3.png!w690x320.jpg[/img]
最经用hp-innowax30*0.25*0.25 测试样品,升温程序为:初始50℃,以3℃/min升到250℃,发现乙醇溶剂峰在10-12min出峰,计算乙醇溶剂峰温度在80-86℃之间,因此,为了避免乙醇溶剂峰,优化升温程序为90℃(保持2min),以3℃/min升到150℃,然后以5℃/min升到200℃,在以10℃升高到250℃(10min)后发现,在10min左右还是有乙醇溶剂峰!!这是为什么呢?2.改用乙腈作为溶剂时,发现同样的现象,不管怎么提高初始温度,总是有乙腈溶剂峰?难道是柱子选的不对?对优化升温程序带来很大的困扰。现在只能在这个时间段关闭质谱检测器,急盼老师指导!
本人按2010版药典中乙醇的气相检测时发现,乙醇的保留时间会相错很多,一般DB-624的色谱柱,乙醇的出峰时间是很快的,一般5分钟左右。然而我最近在作这个检品是有两次却慢的出奇,一次13分钟出乙醇峰,还有一次21分钟才出主峰,这到底是什么原因造成的,请各位专家赐教。
用的FFAP毛细管柱,甲醇为空白溶剂,苯峰在甲醇峰的拖尾处,乙醇与苯峰又很近。当乙醇含量高时又会积分成苯峰。用什么极性的柱子可以分离这三个峰呀。
最近购回一台岛津GC-2010用的是全进样,但是这两天分析时,发现醇峰出得特高!14C上打出来只有3%左右的醇,在2010上醇含量能到20%,[em06] 请大家帮帮忙,不胜感激![em61]
我在做农药残留,跑的三氯杀螨醇,柱子是HP-5,单标出了4个峰,分别在13.806min,16.485min,19.109min,20.485min,对应峰面积分别是6303.8,1840.4,2053.9,1571,我前任以前就选的最后一个峰代表的三氯杀螨醇,那么究竟选哪个峰呢?第一个峰面积大是大,但峰形不好,最后一个太小了,倒是16.485这个峰形也好,峰面积也还可以?究竟是哪个呢?大家又是怎么定的呢?都来说说。
我是按5009做的,单点,结果已经做出来,于是上来参与讨论一下,发现自己做的杂醇油总量要比别人做的小,百思不得其解。后来看到有网友问异戊醇的出峰问题,好些人说只出一个峰,但我历来做的都是两个峰的,异戊醇旁边的峰是活性戊醇。以前用30m的柱子这两个峰是分不开,用CPWAX 57cb 50m的才行,这个柱子的是CNAS专家建议采购的,因为几年前CNAS现场评审考核时候就是因为异戊醇的问题,没有做好。所以现在我看到我做的数比别人的要小,但是不知道怎么报,虽然自己的数是可能对的,但如果有好多数人的结果都是把活性戊醇和异戊醇当成了异戊醇,那我的结果最后肯定是偏离值了。我试了一下,把活性戊醇和异戊醇当一个峰计算,那结果就和其他人一致。 A / B甲醇:0.110 / 0.0944杂醇油:0.198 / 0.180
个人觉得甲醇中的苯系物标样分析中甲醇的峰过大,容易影响至苯的峰,同时实验中可能会因残留的甲醇易干扰采集样品的测试。现那里还有CS2中的七个苯系物标样卖







 我要推广仪器
我要推广仪器
 下载APP
下载APP