乙酰丙酮烯醇式羟基氢的吸收峰为何不是尖峰,而是宽峰?
1 如果想用红外研究不同产地的聚丙烯,需要选择一个PP的特征吸收峰作为参比峰,请问选取哪个峰比较合适?是否这个峰是要吸收强度大,受到干扰的程度小?2 布鲁克红外谱库哪里可以找到?
选用岛津[url=https://insevent.instrument.com.cn/t/Mp]gc[/url] 2010plus,柱子是HP-5的毛细管柱,色谱条件:柱温80度,进样器180度,检测器200度,80度保持5分钟,以20度每分钟的速率升至180度保留3分钟,标准品是正丁醇中乙烯,出来的色谱图,有一个峰型好的峰但是在溶剂峰的后面,新手不会看峰图,乙烯估计多久出峰,恳请大佬们不吝赐教,不胜感激!文档中有图,标品可能因拧开次数多导致峰图和标曲样的图存在较大差异
有人说:在较窄的狭缝下,吸收峰的宽度应该较小,因为吸收峰的表观宽度 应该是入射光的线宽与元素吸收宽度之和. 请大家结合自己的实际工作,来谈谈这种说法是否正确,谢谢!
GB2763中关于亚砜磷的限值要求,标准中指出残留物为亚砜磷 砜吸磷 甲基内吸磷之和,以亚砜磷表示,不知道亚砜磷 砜吸磷 甲基内吸磷这三者之间有什么关系,是可以相互转化,还是同分异构体?
今天,做活性炭管中的三氯乙烯质控样,指控样中的三氯乙烯出峰时间与标准中的三氯乙烯的出峰时间不一样,打进一针纯色谱纯的三氯乙烯,出峰时间还是对不上,是什么原因?望各位老师指点
刚刚接触GPC,做的分析中样品峰是负峰,开始都不敢相信,后来几次调节样品浓度,峰面积大小随之变化,且相关性很好。请教,GPC分析中有样品峰为负峰的情况吗,是什么引起的?
我用丙酮分析纯的作溶剂来配丙烯酸,10000ppm的出峰,峰面积是170万左右,有一些拖尾,然而100-1000ppm的都不出峰,这是吸附太严重了么。总不能配个梯度浓度从10000ppm到100000ppm吧,那也太大了。是不是换个衬管会好些?
问一下 同一个样品,假如在20%乙腈浓度下洗脱下来的峰保留时间是10分钟,之后在50%乙腈下洗脱下来的峰保留时间是20分钟,如果我直接用纯乙腈洗脱,那么这些峰的保留时间还没有没先后次序?
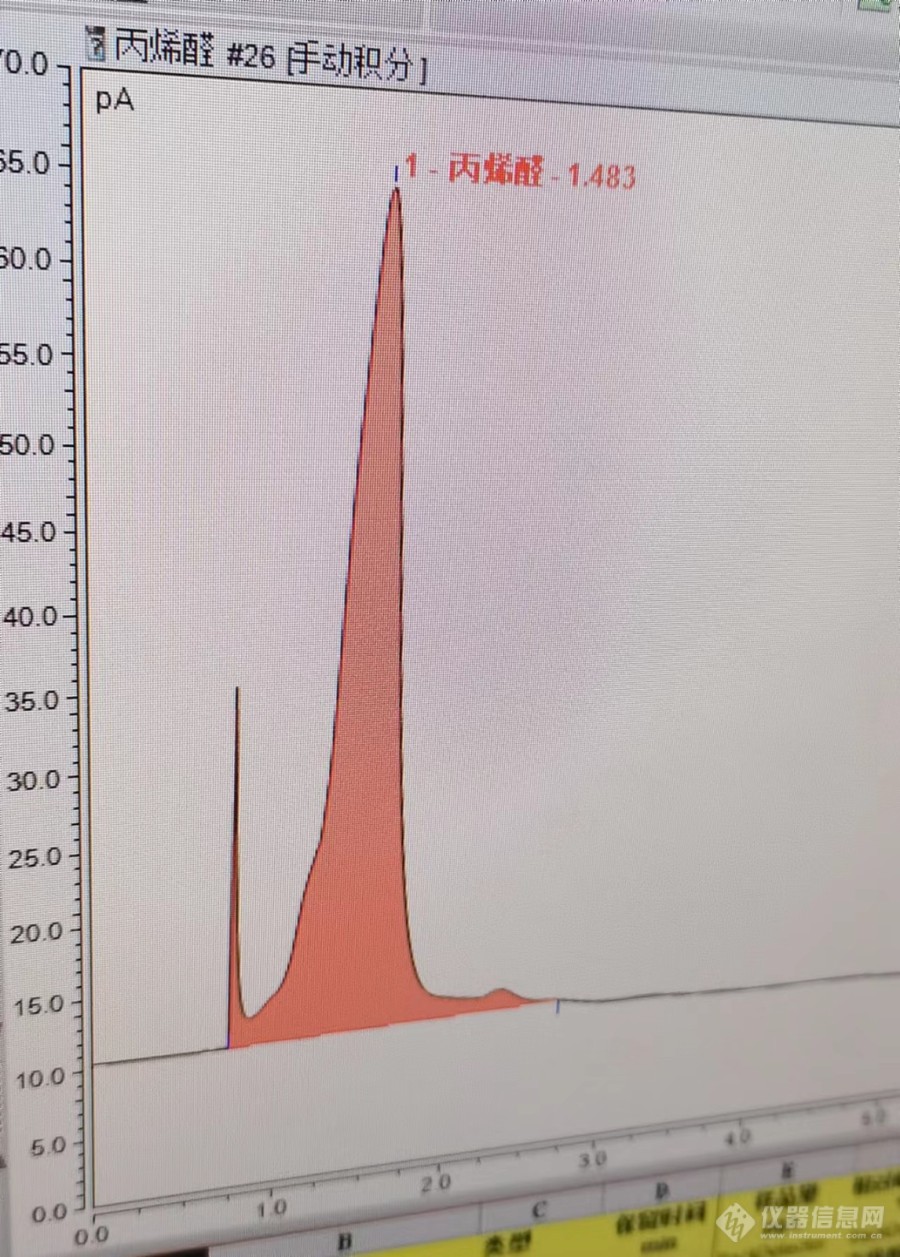
请教大家,样品为水中丙烯醛单标,经吹扫后进入[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url],每个目标峰前面都有这样一个小峰。使用的是WAX柱,30*0.32*0.25,柱子经老化后使用,老化温度220℃。问题一:如何改变色谱条件,能将这个小峰分开?或者提高老化温度,能不能去掉杂峰?问题二:HJ806标准中出锋时间在9分钟,如何将出锋时间延后?[img=,690,963]https://ng1.17img.cn/bbsfiles/images/2023/03/202303281014512066_569_3452300_3.jpg!w690x963.jpg[/img]
今天 刚修完原吸 当查看峰图时发现 峰比以前的峰图平缓 怎么办
各位,近些天,我在做国家标准土壤样品(石墨炉)时,发现一个现象:吸收值用峰高来计算时,吸收值与背景值之比大约为6,但结果却不符合实际的结果,大约相差了10%多。使用峰面积计算样品的吸收值时,吸收值与背景值之比只有1.7—1.8,但结果却与国家标物中心提供的值相符。同时,用峰面积计算标准曲线的吸收值时,溶液浓度可以达到100PPB(吸收值大约是0.4000左右);而用峰高计算吸收值时,标准溶液浓度在100PPB的吸收值已经高达1.2左右了(不知道这样的吸收值有无意义)。我百思不得其解,故此请大家帮助![em04] [em04] [em04]
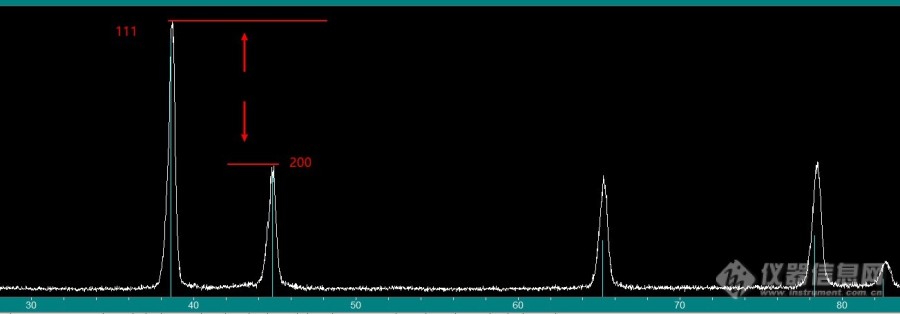
求助各位前辈,我最近对一个Al合金进行了XRD分析,结果如图所示。我的晶粒组织是全等轴晶组织。审稿人认为我的XRD结果反映出合金具有很强的织构,但是EBSD并没有反应出来(根据反极图,织构强度最高为2.0)。我觉得编辑主要针对111峰和200峰,但是我的峰强度和PDF卡片差不多。我知道这种XRD并不能分析织构,但是审稿人问了,希望各位前辈理解。由此,我有以下几个问题,希望能得到各位前辈的解答。1)PDF卡片里的三强峰是如何确定的?为什么111峰本来就比200高?2)测试结果里峰的强度大小和卡片相比的变化是由什么决定的?假如111被测峰强于卡片,是不是说明被测样,至少被测面上,这个方向的晶面很多?3)这种XRD分析方法中峰的强弱能否用于大致判断织构存在与否的一个依据?比如我个人认为,如果200与111峰强度差不多,反倒能说明200织构很强了。[img=,690,240]https://ng1.17img.cn/bbsfiles/images/2022/09/202209182238251618_104_5253538_3.jpg!w690x240.jpg[/img]
请问大虾们,若是怀疑峰的最末尾部分不够纯,怎么样辨别,收集分析呢!既是说如何分段的收集和分析一个怀疑有部分不纯的峰!
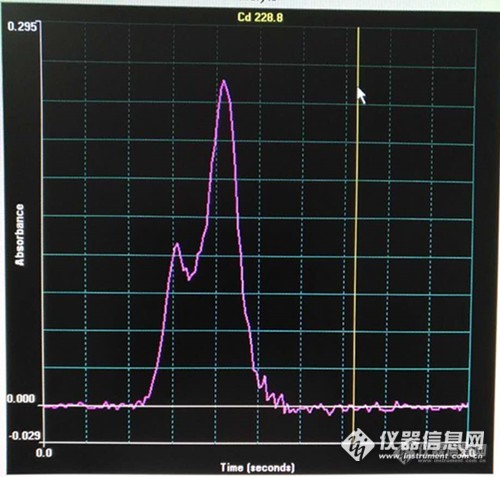
原吸信号的测量方式有两种:峰高和峰面积。今天我在这里不禁要说峰面积好!峰面积好!峰面积好!为何这样说?请参看下图,源自测定实例:http://ng1.17img.cn/bbsfiles/images/2017/02/201702161226_01_2076515_3.jpg图1 http://ng1.17img.cn/bbsfiles/images/2017/02/201702161226_02_2076515_3.jpg图2请看图1和图2这两个峰形图,图1较正常,图2则有分叉,疑似双峰。这两个均是标准物质GSB-6(菠菜)的测定值。若按峰面积测量信号值,则图1、图2中的吸光度值分别为0.0848、0.0898,测定结果为0.141ug/g、0.146ug/g,符合其标值范围0.150±0.025ug/g。若按峰高测量信号值,则图1、图2中的吸光度值分别为0.2684、0.2407,测定结果为0.141ug/g、0.118ug/g,其中图2的结果不能进入符合其标值范围。从峰面积来看,图1和图2差别不大,而从峰高来看,图2因有第二个峰头,显然就损失了不少。就这样我得出了如题的结论。大家怎么看?" alt="
紫外测定含量是不建议选择末端吸收峰作为测定波长,距离终点多大的范围算是末端吸收峰呢
asi19 版友的问题用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](氢焰检测器)检测产品,固定相是GDX-401,甲烷,乙烯和丙烯可以出峰吗?如果出峰,他们的保留时间是丙烯》乙烯》甲烷吗?不知我这样猜想对不对,还请赐教
原子吸收检验铬寻峰时寻到二个峰,杂峰比主峰还要高,是什么原因?
最近再校正硫酸根标准曲线时本来应该在420nm有最大吸收峰的,但是测得时候发现吸光度从450nm到350nm一直增强找不到最大的吸收峰,请教高手这是怎么回事?
请教一个问题:我现在用北京普析通用的[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度计,发现火焰法时燃烧缝上有结晶物,火焰不稳定,燃烧缝上疙疙瘩瘩的,请问各位大虾燃烧缝怎么清洗啊,用什么清洗啊!请赐教!谢谢
问题如题,现在正打算做环丁砜的分析,可条件只是初步知晓, 文章中水水中环丁砜用FID,PEG-20M的色谱柱,不是说水对填料没有影响嘛? 怎么还这样做呢?能说说具体的原因不? 还有就是分析环丁砜中水(可能百分之几十),非要用GDX-403吗? 用毛细柱还是填充柱?谢谢 文章随附件。
今天听同事说起一个问题,就是气相有一天苯乙烯不出峰了,配置大浓度标样也不出峰,然后把柱温升高15℃后,苯乙烯就出峰了。但是出峰时间推后了。。可能是什么原因。
碳氮双键的红外特征吸收峰是多少合成的亚胺类的化合物,想问一下根据红外光谱图怎么样能看出是否生成了碳氮双键,特征吸收峰在什么范围。急,万分感谢。
图一为烯草酮砜的全扫图,图二为烯草酮亚砜的全扫图,但是通过谱库检索检索不到这也两种化合物,还请各位在[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]上有建过这两个化合物的采集方法的能够授业解惑,分享下宝贵方法,谢谢啦[img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111021459230565_8964_3046676_3.png[/img]
各位高人,小妹刚刚学做GC,菜鸟一只,寻求帮助啊~~~~~~仪器是岛津的GC-2014,色谱柱是国产的FFAP,30mm*0.25um*0.32mm 现在分析苯系物,条件设置的是进样口温度是200,检测器温度220,分流比10,柱流速0.6,程序升温40℃保持12分钟,然后每分钟2℃升温至80℃保持2分钟,按道理流速和升温速率已经很低了哎,但是出峰的情况不好,样是甲醇中的9中VOC标样,100ug/ml,进样量为1ul,出峰是溶剂峰,然后甲苯,乙酸丁酯,正十一烷,乙苯和对,间二甲苯是一个峰,(但是明显三个峰头)然后是邻二甲苯,最后苯乙烯。现在的问题是1,苯没有出峰,但是溶剂峰前面有有一个峰,不确定是不是苯峰。2,乙苯和对间分不开啊。特意来寻求帮助,拜托大家帮帮我啊~~~~谢谢啦
最近在看一些文献教材,看到关于[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]燃烧器这一部分。3,燃烧器 试液的细雾滴进入燃烧器,在火焰中经过干燥、熔化、蒸发和离解等过程后,产生大量的基态自由原子及少量的激发态原子、离子和分子。通常要求燃烧器的原子化程度高、火焰稳定、吸收光程长、噪声小等。燃烧器有单缝和三缝两种。燃烧器的缝长和缝宽,应根据所用燃料确定。目前,单缝燃烧器应用最广。 单缝燃烧器产生的火焰较窄,使部分光束在火焰周围通过而未能被吸收,从而使测量灵敏度降低。采用三缝燃烧器,由于缝宽较大,产生的原子蒸气能将光源发出的光束完全包围,外侧缝隙还可以起到屏蔽火焰作用,并避免来自大气的污染物。因此,三缝燃烧器比单缝燃烧器稳定=======================================从来没有见过三缝燃烧器啊?既然三缝燃烧器稳定,那为什么没有大规模普及呢??
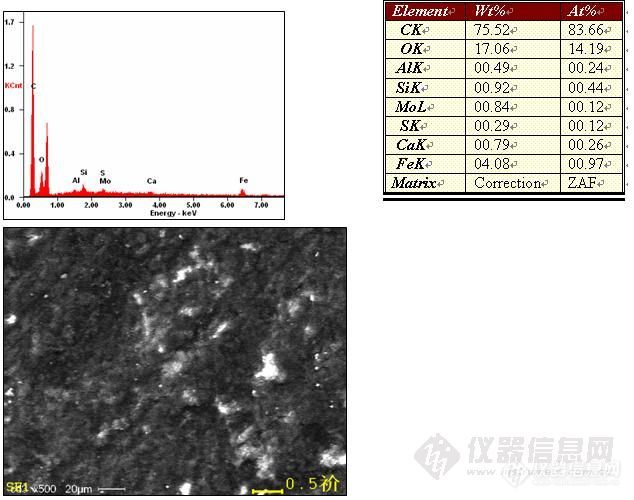
[img]http://ng1.17img.cn/bbsfiles/images/2009/12/200912171346_190507_1614369_3.jpg[/img]低碳低合金钢接箍涂层能谱分析结果见图,涂层目的是防止粘扣。有个疑问,为什么Fe的L系峰比K系的高很多?FK系、MnL系等都在这个位置,结果是这样是不是说明涂层含有F的可能性比较大呢?
用红外光谱分析样品 发现红外吸收峰的半峰宽变大,这说明什么呢?请不吝赐教!
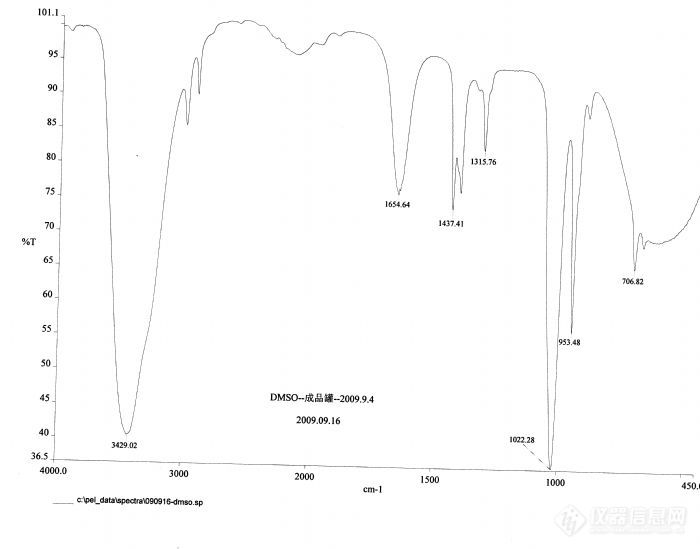
请各位高手帮忙分析一下[size=4]保留时间为4.378(峰号为2)的那个峰是什么东东[/size]?谢谢各位![em09505]第一张图为二甲基亚砜的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]图,面积归一法,第一个为亚砜的主峰,第二个不知道是什么峰,仪器默认为亚砜,第三个是二甲基砜。请大家分析的就是第二个峰。[img]http://ng1.17img.cn/bbsfiles/images/2009/09/200909170913_171497_1765078_3.gif[/img]第二张图为第二个峰(亦即未知峰)的质谱图 第三张图为二甲基亚砜的红外图[img]http://ng1.17img.cn/bbsfiles/images/2009/09/200909170928_171514_1765078_3.jpg[/img]
[em61] 有没有大侠知道色谱分析中BB峰和BV峰中是怎么回事?它们对分析结果有嘛影响?先谢了。。。。。。







 我要推广仪器
我要推广仪器
 下载APP
下载APP