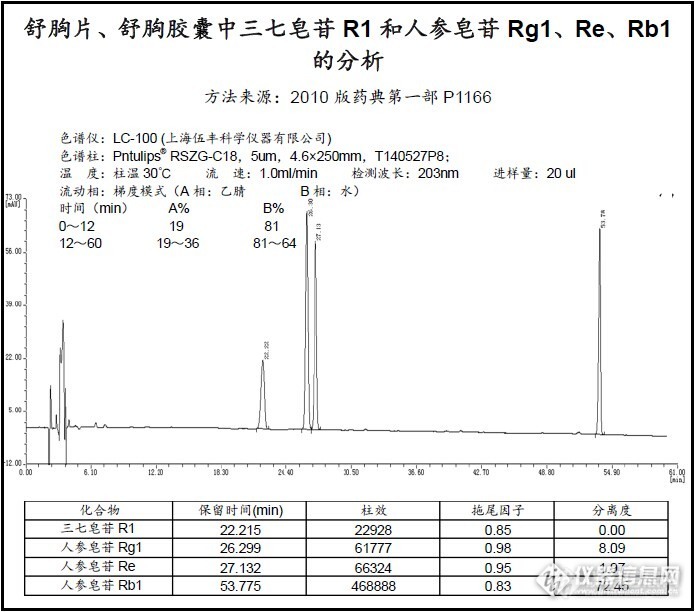
舒胸片、舒胸胶囊中三七皂苷R1和人参皂苷Rg1、Re、Rb1的分析(液相)http://ng1.17img.cn/bbsfiles/images/2014/07/201407151806_506759_2568233_3.jpg
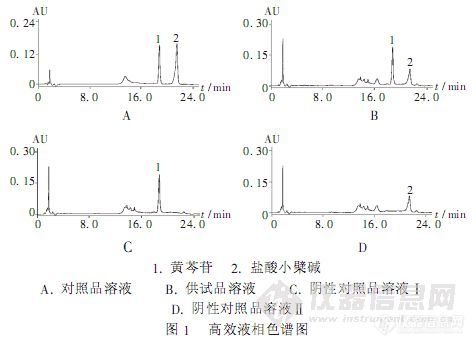
【作者】 彭文; 刘兆雄;【Author】 Peng Wen,Liu Zhaoxiong(Sichuan Deyang Institute for Food and Drug Control,Deyang,Sichuan,China 618000)【机构】 四川省德阳市食品药品检验所;【摘要】 目的同时测定四季三黄片中黄芩苷和盐酸小檗碱的含量。方法采用Diamonsil C18柱(250 mm×4.6 mm,5μm),流动相A为2%的三乙胺溶液(用磷酸调节pH=3.0),流动相B为乙腈,二元梯度洗脱,柱温35℃,检测波长263 nm。结果黄芩苷和盐酸小檗碱进样量分别在0.112 3~2.246 0μg和0.099 2~1.984μg范围内与峰面积呈良好的线性关系,平均加样回收率黄芩苷为99.24%,盐酸小檗碱为99.88%。结论所用方法结果准确,被测溶液稳定性好,可作为四季三黄片中黄芩苷和盐酸小檗碱的含量测定方法。 更多还原http://ng1.17img.cn/bbsfiles/images/2012/08/201208201603_384736_2379123_3.jpg
建立HPLC法测定舒胸分散片中人参皂苷Rg1的含量。方法:以舒胸分散片为研究对象,采用Diamonsil C18(200×4.6mm,5μm)色谱柱,流动相:乙腈∶水(0.05% 磷酸)=25∶75,流速1.0 mL/min,检测波长203 nm,柱温为35℃。结果:人参皂苷Rg1在27.52~440.4μg/mL (r=0.9999)范围内呈良好线性关系;平均回收率为97.1%。结论:本测定方法简便、快速、准确,为舒胸分散片质量评价提供可靠方法。
CCK-8与胸苷结合检测之间是否有相关性?购买的abbkine cck-8检测试剂盒 如何减少由于CCK-8试剂在枪头上或孔壁上的残留所带来的误差?
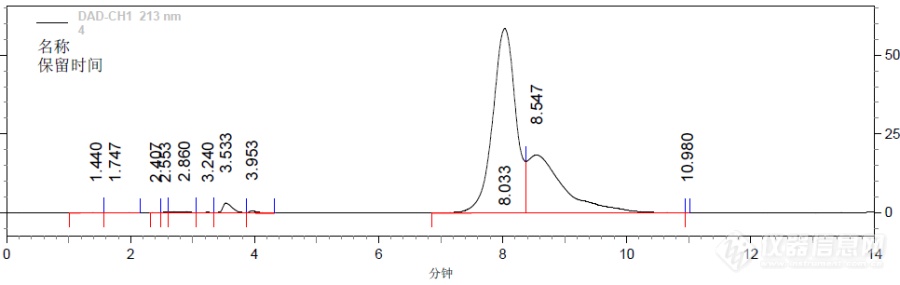
我买的α-熊果苷标品(小公司买的)用C18-AQ做 HPLC检测是2个分不开的峰,而用有机酸柱分析却是一个峰,到底哪个准确? 检测条件如下: C18-AQ:5%甲醇,1ml/min,紫外280nm,25℃。图1: C18-AQ:5%甲醇,1ml/min,紫外280nm,25℃。图2:有机酸柱:5mm硫酸,0.6ml/min,紫外280nm,35℃。[img=,690,218]https://ng1.17img.cn/bbsfiles/images/2019/08/201908091311199678_7583_1750127_3.png!w690x218.jpg[/img][img=,690,299]https://ng1.17img.cn/bbsfiles/images/2019/08/201908091311235804_8826_1750127_3.png!w690x299.jpg[/img]
上海伍丰熊胆川贝口服液中牛磺熊去氧胆酸的测定,请大家多提宝贵意见。
各位老师,跪求浓缩梨汁中熊果苷的提取方法,我们的条件不允许使用SPE小柱,用HPLC-UV检测器定性定量,浓缩梨汁中含有大量的糖类,所以首要考虑的是将糖类出去?跪求各位老师指导,查了一下午的资料,好像也没找到思路,只能在这里求助各位老师了,灰常感谢!
应用高效液相色谱法对熊去氧胆酸原料药进行含量测定。色谱柱为Hypersil ODS25μm , 250mm×4.6mm,流速为1.0mL/min;检测器为UV205nm。该实验有助于对熊去氧胆酸含量测定方法的确定。
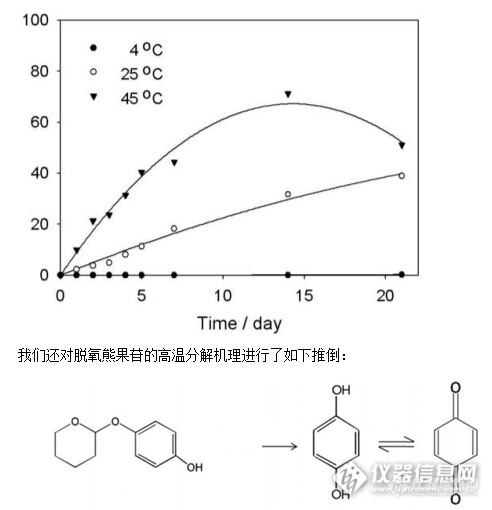
脱氧熊果苷在水溶液中热降解的高效液相色谱法测定 虽然人类的黑色素是皮肤抗紫外线伤害最重要的保障,然而黑色素堆积造成黝黑的皮肤造成了人类美容方面的困扰。黑色素水平的升高也是皮肤疾病,包括黄褐斑,晒斑,和炎症后色素沉着的一大特性。因此,人类越来越渴求一种用于皮肤美容美白兼具治疗作用的产品。酪氨酸是黑色素合成的前体,酪氨酸酶是人皮肤黑素细胞负责酪氨酸转化为黑色素的关键限速酶,通过竞争性抑制剂来降低酪氨酸酶的活性可以降低黑色素在人体黑素细胞内的合成。 经研究许多化合物包括氢醌,熊果苷和脱氧熊果苷能够结合酪氨酸酶的活性位点从而抑制黑色素合成。氢醌是最常规的皮肤增白剂,但其长期应用副作用也较多,包括刺激性皮炎,黑素细胞的破坏,接触性皮炎、褐黄病。熊果苷是熊果植物中一种糖基化的天然的对苯二酚,并且相比氢醌它更安全和较少的细胞毒性,但其在体内研究发现抑制黑色素产生的效率低下,脱氧熊果苷最新被报道是一种新型的皮肤美白剂,具更大的抑制酪氨酸酶活性,并且比对苯二酚和熊果苷更安全。http://ng1.17img.cn/bbsfiles/images/2014/12/201412302146_530363_2165260_3.jpg 熊果苷吸收到皮肤时在会原位产生氢醌,因此,在较高温度下它有潜在不稳定和由于氧化而易于改变其在制剂中颜色。由于脱氧熊果苷是熊果苷的衍生物,故在一些条件下也存在化合物稳定性的问题,这种稳定性问题会导致其在化妆品及医药产品中应用的问题。所以改善其稳定性是其未来应用的一个需要解决的问题。本实验中我们应用高效液相色谱法来分析其在水溶液中的稳定性。并研究了几个影响其降解的温度。材料与仪器:脱氧熊果苷、氢醌、色谱级甲醇、分析级丙二醇、去离子水;紫外可见分光光度计、安捷伦1100、菲罗门C18反相色谱柱、紫外检测波长280nm、流动相甲醇 - 水(60:40(V / V)、进样量20ul、流速1ml/min。结果与讨论:本实验的目的是探讨脱氧熊果苷在溶液中的热稳定性,所以我们首先确定了其溶解度及水溶液的紫外吸收图谱,其后建立了HPLC方法定量脱氧熊果苷,对熊果苷的热降解动力学进行了分析。脱氧熊果苷水溶液的制备--因为去除了葡萄糖侧链的羟基基团,脱氧熊果苷在室温下难溶于水,故采用丙二醇助溶,可将脱氧熊果苷的溶解度在丙二醇及丁二醇的助溶下达到13%(W / W)。美国食品和药物管理局(FDA)已经确定丙二醇是一种安全的成分可应用在化妆品、食品及药品中;世界卫生组织(WHO)也确定了它是安全可使用的。像水一样应用普遍的丙二醇常作为溶剂或湿润剂应用于化妆品中还有助溶的作用。虽然乙醇也可以起到助溶的作用,但考虑到其对皮肤的刺激性,我们采用了丙二醇作为脱氧熊果苷在水中的助溶剂。脱氧熊果苷的紫外吸收图谱:为确立脱氧熊果苷的紫外吸收情况,采用紫外可见风光光度计收集脱氧熊果苷水溶液的紫外吸收图谱,采用0.05 和 0.1 mM的脱氧熊果苷去离子水溶液(含10%的丙二醇),于石英池中200-400nm下进行测定。结果如下图:显示一个最小的248nm和两个232和283 nm的最大值。吸光度水平随浓度的增加而增加。http://ng1.17img.cn/bbsfiles/images/2014/12/201412302147_530364_2165260_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412302147_530365_2165260_3.jpg标准曲线浓度范围各为12-144mg/升,R2大于0.995。(其中下面为脱氧熊果酸,上面为氢琨)http://ng1.17img.cn/bbsfiles/images/2014/12/201412302148_530366_2165260_3.jpghttp://ng1.17img.cn/bbsfiles/imag
亚洲动物基金中国高级外科兽医莫妮卡透露,在该机构曾经救治过的277头黑熊中,没有一只黑熊的胆囊是正常的。165头被使用无管引流术取胆的熊,99%的患有胆囊炎,一半以上患有胆囊息肉等。还有很多黑熊死于肝癌。
什么是赶酸,具体是怎么赶酸的?
[b][size=18px][color=#06948c]胸腹部的脾经大药房——不给疾病任何藏身之地[/color][/size][/b]脾经还有好多穴位都在腹部上,一推腹就全给推了。[b]1.推腹法[/b]脾经有许多穴位分布在腹部,[b]通过推腹可以一次性刺激这些穴位[/b]。它们通常位于人体中线旁开4寸的位置上,如果在这个位置上有痛点,可能是脾经的问题。[b]2.按揉拍打大包穴[/b]大包穴位于侧胸部,腋中线上,第6肋间隙处,也是脾脏的最后一个穴位。[b]主治疾病:[/b]胸部肋骨疼痛、气喘等。[b]按摩方法:[/b]用右手食指和中指指腹点按左侧大包穴,揉动脾经,使穴位产生明显的酸、麻、重、胀感。最后用掌心轻揉,轻轻拍打腋下放松。操作时要左右两侧交替进行,按揉10-15分钟,每天1-2次。
益母草与川芎是中药成方制剂中配伍应用频率较高的药对,益母草主要含水苏碱等生物碱类活性成分,川芎主要含阿魏酸等活性成分。对益母草药材及其制剂《中国药典》常以雷氏盐剩余比色法测定益母草总碱含量。在对含益母草及川芎这一药对的制剂进行质量控制时,由于川芎所含生物碱也能与雷氏盐反应生成沉淀,且雷氏盐剩余比色法本身重现性较差,故不宜采用该法。为此,本实验建立了以薄层扫描法测定益母草与川芎合煎液中盐酸水苏碱含量的方法,为制订含有益母草与川芎药对制剂的质量标准提供参考。 1 仪器与试药 CS-9000型双波长薄层扫描仪(日本岛津):939薄层制板器(重庆南岸贝尔德仪器技术厂);定量毛细管(Drummond USA)。盐酸水苏碱对照品(中国药品生物制品检定所,712200105);益母草、川芎(购于安徽毫州)。硅胶G(化学纯,青岛市北区海化干燥剂厂);强酸性阳离子交换树脂(732型,上海化学试剂采购供应站);其余试剂均为分析纯。 2 方法与结果 2.1 益母草与川芎合煎液的制备 取药材100g(益母草-川芎:1:1),加水煎煮2次,每次2h,每次加12倍量水,合并煎液,滤过,滤液浓缩并定容至100mL,即得合煎液样品。 2.2 对照品溶液的制备 取105℃干燥至恒重的盐酸水苏碱对照品适量,精密称定,加乙醇溶解定容,制成1.2mg/mL的对照品溶液。 2.3 供试品溶液的制备 取合煎液样品适量,离心(4O00r/min)5min后,精密量取上清液lmL置小烧杯中,加蒸馏水1OmL,用稀盐酸调pH 1~2,通过已处理好的强酸性阳离子交换树脂柱(内径1cm,长1Ocm)用水洗至流出液无色,弃去水液,再以乙醇-氨水(8:2)150mL洗脱,收集洗脱液,蒸干,残渣加乙醇溶解,定量转移并定容于lmL容量瓶中,摇匀,作为供试品溶液。 2.4 薄层色谱与扫描条件 吸附剂:硅胶G-O.5%CMC-Na薄层板(厚约0.5mm,105℃活化lh,置干燥器中备用):展开剂:丙酮可-无水乙醇-盐酸(10:6:1) :显色剂:喷以改良碘化铋钾-1%FeC13无水乙醇液(5:1)。,冷风吹至斑点显色清晰。 描方式:双波长反射式锯齿扫描:检测波长:λs=525nm,λR=660nm:狭缝:0.4mm×0.4mm: 线形参数SX=3。 2.5 标准曲线的制备 分别精密吸取盐酸水苏碱对照品溶液4.0、6.0、8.0、10.0、12.0μL,分别点于同一薄层板 ,依上述条件展丌,显色,在薄层板上覆盖同样大小的玻璃板,周围用胶布吲定,然后扫描测定各斑点的峰面积积分值。以峰面积积分值(A)对点样量(C)进行回归,得回归方程:A=-37152.7+7579.18C‘r=O.9986。表明在4.8~14.4μg范围内盐酸水苏碱斑点峰面积积分值与点样量呈良好的线性关系。 2.6 稳定性考察 取供试品溶液5μL点样,依上述条件展开,显色,在1h内每隔10min扫描测定1次,结果斑点峰面积积分值RSD为4.6%(n=6),表明斑点在显色后1h内基本稳定。 2.7 精密度考察 精密吸取供试品溶液在同一薄层板上点相同量5点,每点5μL,按上述条件展开,显色,扫描测定,斑点峰面积秋分值RSD为1.33%(n=5),对其中一点连续扫描测定5次,斑点峰面积积分值RSD为0.62%(n=5),表明同板精密度和仪器精密度较好。 2.8 异板精密度试验 取薄层板3块,在每1块薄层板二分别点对照品溶液4、6μL及供试品溶液5μL,依法展开,显色,扫描测定,采用外标两点法计算含量。结果测得供试品溶液中水苏碱含量为1.1308 mg/mL(RSD=3.01%)。 2.9 回收率测定 精密量取已知含量的合煎液样品0.5mL,加入盐酸水苏碱对照品溶液0.5mL,混匀后,按2.3项方法制备供试品溶液。精密吸取供试液5μL,对照品溶液4、6μL,变又点样于同一薄层板上,按上述条件展开、显色、扫描测定,用外标两点法计算。 2.10 样品测定 取合煎液样品,按2.3项 方法制备供试品溶液。精密吸取供试品溶液5μL,对照品溶液4、6μL,分别交叉点于同一薄层板上,按上述条件展开,显色,扫描测定,用外标两点法计算样品中盐酸水苏碱的含量。 3 讨论 在供试品溶液制备中,以甲醇-氨水(8:2) 或乙醇-氨水(8:2) 作洗脱剂,提纯效果均较好。因乙醇比甲醇价廉且安全,建议采用乙醇-氨水(8:2)作为洗脱剂。本实验对洗脱剂用量考察结果表明,用14OmL即可将盐酸水苏碱洗脱完全。为了保证洗脱充分,本实验中确定用150mL洗脱。 盐酸水苏碱为水溶件物质,在薄层板上显色时受湿度影响很大。通过实验考察,优选出其较好的显色方式为:展开后,取出,晾干,丁105℃烘lOmin,再喷显色剂,冷风吹至斑点色清晰。 益母草有效成分水苏碱含量较低且波动大,容易造成含益母草的制剂有效成分低、产品质量不稳定 ,应增加水苏碱含量控制项目。本实验建立的含量测定方法可为制订含益母草及川芎制剂中水苏碱的定量标准提供参考。参考文献:[1]国家药典委员会.中国药典(一部)[S].北京:化学工业出版社,2000.237,455,564.[2]张玲,时延增,于宗渊,等.舣波长薄层扫描法测定益母草LJ服液中水苏碱的含量[J].中国药科大学学报,1996,27(1):16—18.[3]张玲, 宗渊,李国宝,等.7种含益母草中成药中水苏碱的含量测定[J].药物分析杂志,1996,16(3):181—183.
食品中的汞,一般都用微波消解,湿法消解也行,常用的还是微波消解。但是消解完之后到底赶不赶酸呢,赶酸温度不能太高,这样时间就会拉长;不赶酸又担心酸度太大,对检测结果又影响。那是赶酸还是不赶酸呢,大家做过对比么?
地表水标准要求做砷汞硒都需要消解,但是标准里都没有提到赶酸的问题,我们更换了新的cem消解仪,咨询工程师,说是不需要赶酸,但是之前的同事在做的时候有赶酸这一步,对这个问题心存疑虑,不知道这样消解出来的样品是否还需要赶酸,如果赶酸,应该怎么赶?还需要买赶酸的仪器吗?
[color=#444444]查了文献,大多都是用甲醇溶解后进液相色谱,说是溶液稳定性良好。[/color][color=#444444]但是我自己做的时候,有甲醇存在于稀释液中,溶液就不稳定,两个小时就会有熊去氧胆酸甲酯产生。。[/color]
大家讨论下样品前处理的赶酸这一步骤吧,本人感觉赶酸也很重要。赶酸温度设高了,被测元素可能会挥发损失掉,温度设低了,赶酸耗时。习惯上,我都是赶酸赶到还剩1毫升,然后定容到10毫升,再吸1毫升,定容到10毫升本人做砷的时候,加热板温度设200度(实际温度不知道多少,比200度低一点的吧),做过一次回收,没啥问题做锑,赶酸设了180度,做了回收,也没啥问题,(下次我再试试200度吧)做硒,180度,也没啥问题做锡,赶酸我设了150度,也没啥问题,但后来又做了一次,回收却不行,(我不知道问题出不出在赶酸这一步,我测锡荧光值也不稳定)当然,做砷啊等比较好测的元素,赶酸貌似不是特别重要。本人也是新手,前辈们指教
做盐酸川芎嗪氯化钠注射液时(标准号为WS1-(X-113)-2003Z)检不出有关物质流动相:1%醋酸钠溶液-甲醇波长302(最大紫外吸收296)我不知道为什么要选择302主峰出来很漂亮,理论板数、托尾因子都符合规定不知道是没有杂质还是标准有问题(原料药不检有关)包栝到市面上买回来的样品出检不出有关物质请教大家!给点意见!
大神们,赶完酸颜色不均匀,是因为赶酸中间发生了什么反应吗
我最近在做化妆品重金属测试。测铅砷汞,我先问下我测砷的时候我在赶酸前加了1mol/L的硫酸5ml,我想问下那我赶酸的温度要多少才能赶酸,铅 汞赶酸的温度多少菜合适,我用的试剂是4HF 3HNO3
分离黄芩苷 绿原酸 连翘苷的一些问题液质联用分离黄芩苷 绿原酸 连翘苷 先用的液相色谱摸索流动相及流动相的比例什么的 最后选用乙腈和乙酸胺 但试过之后 发现峰分的不太开 采用梯度洗脱效果也不明显 感觉有两种物质的峰重合了 因为只得到俩峰 柱子是2.1*150 的 流速是0.2μg/min 想请问 如何才可以较好的分离 是流动相的问题么?那应该选什么流动相呢?柱子不能变 只有2.1*150 的
消解还有硅材质的样品,通常都会用到氢氟酸,大家是怎么赶酸的。因为要测试B元素,所有没有用硼酸,用电热板赶酸,大家是怎么样来操作的,用什么容器,赶酸温度是多少?最好有图片发上来分析一下。
赶酸是赶高氯酸还是硝酸还是硫酸?哪个对砷结果有影响
样品消解后赶酸什么方法比较好呢蒸干 感觉时间长 有没有什么号的方法呢微波消解后怎么赶酸
我测的是玉米中的cd,用高氯酸和硝酸混合消解,请问是在180度下赶酸至2-3ml(这时管内有一团白色烟雾,在回旋)还是在150度,赶至1-2ml,再加纯水继续消解,反复2次,赶的干净呢?
大家微波消解后是否赶酸?用的赶酸设备都是什么牌子的?效果如何?欢迎大家上图分享经验(有加分哦)
在进行仪器测定之前,我们一般都需赶酸,消解罐直接赶酸至近干,好像不太好目测,消解罐又长又细,里面的液面看不太清楚,大家是怎样判断的呢?
最近遇到问题,就是做大米镉的质控样,按照方法先微波消解,然后140℃赶酸至近干,最后定容后上机测定,结果显示结果偏低不少。问题就是可不可以不用赶酸直接上机呢?大家在上机的溶液一般酸度多大呢?
石墨炉测定,一般微波消解后都需要赶酸。赶酸的操作可谓八仙过海各显神通,有用赶酸器通风柜内赶、电热板直接赶、转移后赶、水浴赶,还有赶酸器收集酸气通过碱液中和成水的装置。
很多方法中,都存在赶酸的问题,我想问问大家为什么要进行赶酸呢?赶酸是用什么原理呢?沸点还是其他性质呢?







 我要推广仪器
我要推广仪器
 下载APP
下载APP