哪位老师用GB23200.121法测过呋虫胺、氰霜唑、环丙唑醇,定量限附近做不出来,比标准高了10倍
哪位老师用GB23200.121法测过??呋虫胺、氰霜唑、环丙唑醇,定量限附近做不出来,比标准高了10倍
环己六醇如何作鉴别?环己六醇做鉴别怎么做?原理是什么呢?
流动相是甲醇, 甲酸铵(2mmol/L)+0.01%甲酸水,但是环丙唑醇是两个峰,怎么回事呢?[img]https://ng1.17img.cn/bbsfiles/images/2023/09/202309191544148736_6337_5346633_3.png[/img]
流动相是甲醇, 甲酸铵(2mmol/L)+0.01%甲酸水,但是环丙唑醇是两个峰,怎么回事呢?[img]https://ng1.17img.cn/bbsfiles/images/2023/09/202309200708288399_5763_5346633_3.png[/img]
要测定环己醇和环己酮的校正因子?该用什么做内标物比较好?对柱温等其他温度该怎样设定比较好?另外麻烦高手教一下怎样设置程序升温啊
[color=#444444]双氧水做氧化剂,杂多酸做催化剂氧化环己醇,反应产物主要是环己酮([url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析)外,还有己二酸,除此之外还有什么产物?越具体越好,分别用用什么方法定性定量分析?[/color]
美国输日红加仑子检测出丙环唑超标輸入時のモニタリング検査の結果、米国産生鮮レッドカラントから基準値を超えるプロピコナゾール(PROPICONAZOLE,用途:殺菌剤 基準値:0.05ppm)が検出されたと発表した。この結果を受け、米国産レッドカラントのモニタリング検査の頻度を30%に引き上げると通知。
想用外标法测甲醇和环氧氯丙烷混合物,怎么弄啊,我之前就是自己看书学的,我是这样做的自己先配置甲醇和环氯的溶液,二者的质量我都知道,然后就进样求校正因子嘛~我配置三组不同质量分数的甲醇和环氯,测出来的校正因子都不一样。是我理解错了吗就是在输入方法的时候,里面有个浓度的数值,我输入的就是我称的样品的质量,这里的浓度指的是摩尔浓度吗,而且出来的含量指的是什么,怎么计算呢?想聊的加我Q,拜托了~
[color=#444444]环己烯水合制备环己醇的实验,反应结束后用乙酸乙酯萃取,最终得到含有环己烯、环己醇、乙酸乙酯的有机相(应该有一些副产物)。[/color][color=#444444] [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析产物中环己烯和环己醇的质量,来确定环己烯的转化率和反应选择性。准备用内标法,没确定好用什么内标物,最好是毒性比较小的,望大神相助!谢谢![/color]
以下所述校正因子均为FID中物质相对正庚烷的相对校正因子! 查资料环己酮的FID校正因子为1.38,但是环己醇仅有TCD的校正因子,无FID的校正因子。 若按照有效碳数法计算,查资料羰基贡献为0,即环己酮校正因子按有效碳数法计算:7*98/(100*5)=1.37。请问下羟基的贡献是多少呢,环己醇FID校正因子是多少? 文献理论上看,羰基贡献为0,羟基贡献应大于零,也就是说环己醇的响应因子应比环己酮大,也就是环己醇的校正因子比环己酮小。 仪器实测环己酮校正因子1.20,环己醇1.11。 求帮助!
用Wax毛细管柱测定乙醇、二氯甲烷,柱温、进样口温度、检测器温度需用什么温度 溶液直接进样,二氧六环作溶剂
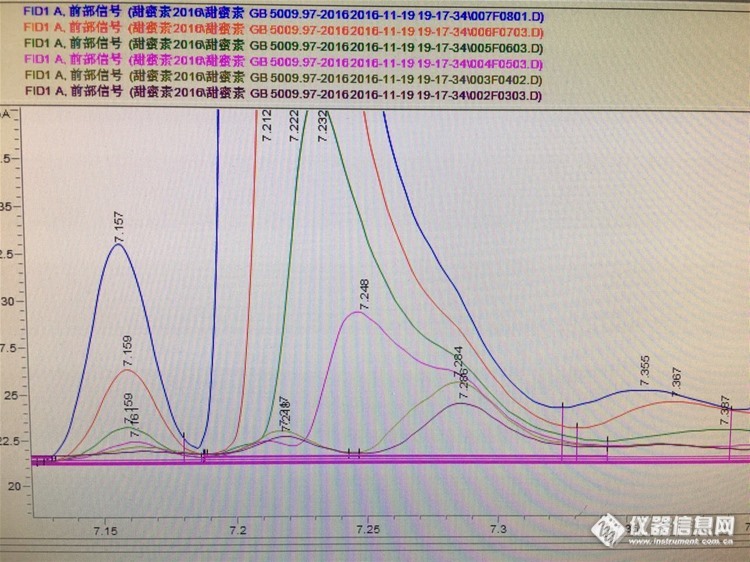
请问有人做GB 5009.97-2016的甜蜜时光素吗?为什么浓度低的环己醇的峰很奇怪……好友回复:进的标品的话,那就只能舍弃这个点了,这不关浓度的事,因为各物质都存在响应的高低,既然标品都成这样了,如果配置过程中没问题,那就是这个浓度不行了,你先再老化一下柱子吧,你重新割一下柱子,并且确保切口平整,柱子伸进进样口最好5mm这样,先从高浓度开始进样,试试看吧大家说说看~http://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_669708_3150883_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016112217295387_01_3150883_3.jpg
我采用的是甲醇:水96:4,或者甲醇:水75:25做流动相,检测波长223nm,测原药的时候没有问题,但是试样(250g/L丙环唑乳油)却总是分不开。[em0809] 我做液谱分析还太少,请达人帮忙分析分析
【问题】有做酒精中甲醇的吗?最近甲醇峰与叔戊醇内标峰,柱效都下降的快,峰又肥又矮,试过换衬管,老化柱,换柱,均没效(innowax的柱子,fid测其他物质没毛病)【回复】调下尾吹,氮气、氢氢和氧气流量比例要最佳,如果不行,在调节下分流比,调大点试试
求助:工业级环己醇测定的相关国标
用0.1%磷酸做缓冲溶液 与甲醇(0.3:0.3)做流动相,基线很不平,向下漂移,请问是什么原因呢?(平衡了两个小时。)
以草酸与1,2-环己二醇为原料缩聚制得聚乙二酸环己二醇酯,以甲苯为溶剂,室温(25摄氏度)条件下测其产物粘度。哪位大神能帮忙找找该条件下聚乙二酸环己二醇酯粘度的K值和a值?或者找类似的物质如聚己二酸环己二醇酯在该条件下的K值和a值代替也可。
液相加不加缓冲盐以及流动相的保留问题各位专家,我用反向C18柱做的是对羟基苯甲酸酯的检测,想用甲醇水做流动相,看文献上说可以用乙腈水、或者用甲醇与缓冲盐溶液做流动相,我想问一下,在这里是否需要加缓冲盐,不加行不行?另外,如果我的样品里含有少量乙醇和异丁醇,会不会在色谱图上出峰?大家见笑了,因为是初学者,很多问题都不懂,也不敢贸然去试验单位里刚刚买的进口机器,所以先请教大家一下,非常感谢您的指导!
请问各位:有谁做过环戊酮氰醇的GC样品分析吗???我目前分析的结果是环戊酮氰醇与原料环戊酮的GC中的比例一直不稳定,似乎在GC中环戊酮氰醇会转化为环戊酮,请问各位有什么好的方法???
想测量一下环体中十八醇的含量,进样条件该怎么设置呢?
请问苯醚甲环唑,溴螨酯,吡虫啉,啶虫脒是用什么来溶解配标液的 啊?我们实验室只有正己烷和乙腈是色谱纯的。。还有大家异狄氏剂,三氯杀螨醇,用GC测出来是一个峰还是多个峰的 啊?色谱程序改变同一标准农药它的峰个数都不一样啊、是因为带入杂质了吗?
我在用VARIAN 的PAH柱做动植物油中16种多环芳烃的实验,请问PAH柱和C18柱填料差不多么?能走纯水么?为什么我走样 越进样压力越高?要是走纯乙腈就没事,水的比例一大压力就好高,求助啦!谢谢!!!!
最近在做四环素类药物,开始液相做的,磷酸盐缓冲液流动相不行,某一个国标的,后来换草酸,出峰和响应均能达到要求,不过实际样品处理的时候回收率不好,前处理是1%高氯酸提取,重复,过C18小柱,也是国标处理方法,纯标准品过柱回收率还可以。液质目前问题是响应不好,仪器是安的6410,只能到50ppb,流动相是0.3%甲酸水和0.3%甲酸甲醇。做的好的达人请指点,多谢

昨天有版友问我,聚乙二醇6000的环氧乙烷和二氧六环未出峰,用的是elite-1的柱子。顶空平衡45分钟,针温是100度,炉温70度,传输线105。操作规程按照药典。可能的因素有哪些。有没有做过这个项目的老师回答一下。
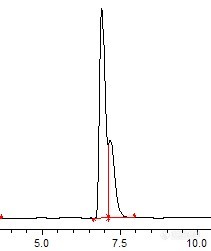
http://ng1.17img.cn/bbsfiles/images/2011/02/201102241706_279281_1928138_3.jpg我用的岛津的液相检测丙环唑,C18柱,流动相是甲醇+水=90:10,波长220nm,出的是双峰,但是连在一起,不知道有没有问题,不知道大家做是不是这样?
R-3-奎宁环醇厂家提供色谱柱为β-DEX 120的方法,目前手头上有的相似固定相Cyclosil-B色谱柱,检测不出峰,想问下大家都是如何检测的?
之前在芒果中测苯醚甲环唑,做加标回收率试验,加标样品出来的结果,比纯标准品和用基质配制的标准品出来的结果都高很多,这是什么原因?如果说是基质效应的话,那为什么用基质配制的标准品也低于加标的样品呢?求大侠解惑!前处理用的QuEChERSGC-ECD检测
有谁做过环己烯的纯化呀,请指导一下,非常感谢!
新手做薄层,想用薄层色谱法分离蒽醌类及其苷类成分,之前用的是二次展开,后来摸索出一个氯仿-甲醇体系能够实现一次展开,但体系中氯仿的毒性太大,因为是想要做欧洲药典方面的内容,所以不建议使用毒性大的成分。之后尝试过二氯甲烷替换氯仿,条带不清晰且扭曲,想询问一下有什么别的体系可以替换吗?







 我要推广仪器
我要推广仪器
 下载APP
下载APP