[em09511] 谁有乙酰肼的分析方法 能不能分享下 谢谢
乙酰氨基葡萄糖糖精乙酸脂能衍生吗?江湖救急啊!!!急急急
我们公司做毒死蜱,合成工艺第一步是三氯乙酰氯和丙烯腈加成氯苯为溶剂,要根据标样的归一情况来确定投料量。标样里面丙烯腈大约10%三氯乙酰氯3.5%氯苯85%左右,之前一直是这个数据,今天突然三氯乙酰氯就变成2.8左右了。色谱条件是,进样口260,柱温90,检测器260。载气氮气,氢火焰检测器,柱子是SE—30非极性柱,用的空气发生器。仪器是国产的福利GC9790②。还有个问题就是氯苯有时候会出平顶峰,进样量0.2微升不到都平顶。以上两个问题还请各位大侠指导一下。
各位大侠,小弟欲请教下有没有用瓦里安2200离子阱气质检测甲胺磷和乙酰甲胺磷,急求方法,我这两天一直在做但是全扫效果都很差,欢迎赐教!谢谢
最近做乙酰甲胺磷发现一个奇特的现象。方法如下,乙腈提取提取,浓缩后过某大品牌氨基柱,乙腈甲苯3:1淋洗,浓缩后甲醇水定容,上LCMSMS,定量用基质标。基质为普通白菜。上机试液理论值100ppb,结果乙酰甲胺磷没有出来,开始以为氨基柱的问题,后来一个突发想法,把试样稀释了十倍,再上机分析,乙酰甲胺磷回来了,而且值为6~8ppb,符合回收率。LCMSMS为watersuplc,TQD,C18液相柱。请问各位同行,有用液质做乙酰甲胺磷的大侠吗?基质效应那么大?
邻氯乙酰乙酰苯胺的国标或行标
乙酰甲胺磷经过S、A、W等品牌的SPE柱子净化后,回收率能有多少?A、W的NH2柱乙腈甲苯(3+1),回收率为0;W的工程师在W实验室得到同样的结果,用乙腈甲醇(3+1),回收率为0;乙腈水(3+1)回收率为114%您,怎么看?
各位大侠,我们在实际操作中,会偶尔遇到有的样品本身背景颜色过滤不掉,以致加入显色剂后的显色效果不是很好,我们已经试过很多方法都过滤不掉背景颜色的,还有哪些会对乙酰丙酮显色有干扰,谢谢
最近在做乳品的扩项,左旋肉碱测定用了新发布的GB 29989.2013,里面用到乙酰肉碱转移酶。原理中是这样讲的“左旋肉碱与乙酰辅酶A 在乙酰肉碱转移酶的催化下反应生成乙酰肉碱和游离的辅酶A。”操作部分只说“乙酰肉碱转移酶(CAT)溶液:吸取100 μL 乙酰肉碱转移酶悬浮液,经1500 r/min 离心10 min,弃去上层清液,沉淀用2 mL 水溶解。临用时配制。”文中没有提到乙酰肉碱转移酶的浓度信息。因为现在要订购试剂,不知道买多少合适,酶又都很贵。是说这个酶不溶于水吗?作为催化剂的话,是不是要一点点就够了?
求乙酰肼,恶二唑酮,唑丙酮,三嗪酰胺检测方法,跪求,谢谢
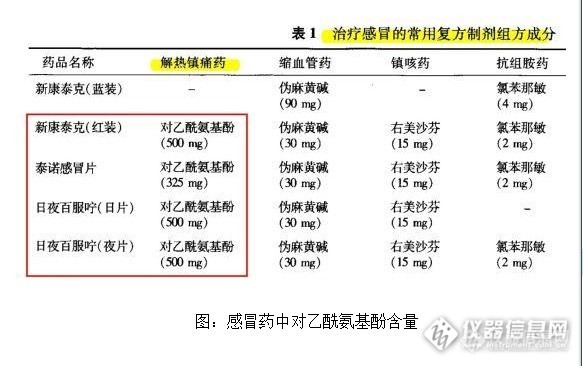
点击查看FDA声明原文 FDA警告,过量服用对乙酰氨基酚药物将会导致肝功能衰竭甚至死亡,高危人群包括在24小时内服用超过处方规定剂量药物的患者,同时服用超过一种含对乙酰氨基酚成分药物的患者,以及在服用含对乙酰氨基酚成分药物时饮用含酒精饮料的患者。 FDA自2011年起开始要求制药企业将对乙酰氨基酚的含量控制在每单位325毫克以内,但截止至2014年1月14日,市场上仍有部分单位对乙酰氨基酚含量超过325毫克的药物正在流通销售。 FDA表示,将在近期启动相关法律程序,禁止开具每单位对乙酰氨基酚含量超过325毫克的药物处方。 国内康泰克、百服宁等超标近54% 调查发现,国内多家知名品牌感冒药的对乙酰氨基酚含量高于每单位325毫克,包括新康泰克、百服宁等。 中美史克生产的新康泰克美扑伪麻片(红装),其每片对乙酰氨基酚含量同样是500毫克。 上海施贵宝制药有限公司生产的日夜百服咛、加合百服宁的每单位对乙酰氨基酚含量为500毫克,超出美国FDA规定含量上限54%。 此外,有多个品牌感冒药的单位对乙酰氨基酚含量达到美FDA规定的上限325毫克,它们分别是:白加黑、泰诺、银得菲。 其他品牌如快克、感康、康必得等单位对乙酰氨基酚含量在250毫克以内。 走访沪上多家药房后发现,最为常见的白加黑、新康泰克等感冒药均为处方药,需要处方及身份证才能购买。 大量非处方药含对乙酰氨基酚 不过,一药房工作人员提醒道,前述感冒药被转为处方药管理,并非缘于对乙酰氨基酚的过量,而是系其含有伪麻黄碱的缘故。 事实上,由于良好的镇痛祛热疗效,对乙酰氨基酚药物使用普及度很高,国内亦将其列为非处方药(OTC),即不需要持有医生处方即能买到,同时也能通过网络合法销售。 搜索淘宝等线上商城后发现,在网上不仅可以随意买到各种各样品牌的对乙酰氨基酚药品,而且购买数量亦完全没有限制。 沪上一医药销售人员表示,由于在各种药物中使用广泛,以及非处方药可以任意购买的缘故,患者容易同时购买多种含有对乙酰氨基酚的药物,导致患者在不知情的情况下过量服用。 根据FDA统计,在1998年至2003年间,对乙酰氨基酚过量服用是导致病人肝衰竭的主要原因。美国疾病控制中心在2007年也曾经发布报告称,全美每年有1600起急性肝功能衰竭,其中对乙酰氨基酚过量服用是最大缘由。 该销售人员解释道,美FDA长期以来都对药物中对乙酰氨基酚的含量作出规定,但因为非处方药的关系,仍有大量患者过量服用,所以FDA将对乙酰氨基酚的含量限制在325毫克以内,也是为了在不影响疗效的情况下尽可能的让患者减少服用,降低副作用。 FDA在此次声明中亦表示,将采取新的行动针对对乙酰氨基酚类的非处方药进行监管。 小知识:对乙酰氨基酚(扑热息痛) 对乙酰氨基酚是乙酰苯胺类解热镇痛药,别名有乙酰氨基酚、扑热息痛、醋氨酚、退热净等。该药具有解热镇痛作用,主要用于缓解轻、中度的疼痛,如关节痛、头痛、神经痛、牙痛及痛经等。 作为非处方药,虽然有质量稳定,疗效确切的优点,但该药的滥用,使用不当也会产生不良反应或严重的肝毒性和肾毒性。 此药对肝损害是最主要的不良反应,其次是引起肾衰,此外也可能导致血小板减少、哮喘发作等症状。 在无其他药物或无酒精干扰情况下,对乙酰氨基酚的安全剂量为:成人口服每次300-600毫克,最大日剂量不超过4000毫克,退热疗程不宜超过3天,镇痛疗程不宜超过5天。儿童12岁以下按体重每次10-15mg/kg,疗程不宜超过3天。 若患者饮酒或空腹或与其他药物有相互作用的药物合用时,应调低剂量或禁用。

乙酰基六肽-8,别名阿基瑞林,是一种优质的祛皱化妆品原料, 其抗皱活性高, 副作用小,已在各高端化妆品系列中应用。【详情请咨询国肽生物】它能局部阻断神经传递肌肉收缩讯息,影响皮囊神经传导,使脸部肌肉放松,达到平抚动态纹、静态纹及细纹;有效重新组织胶原弹力,可以增加弹力蛋白的活性,使脸部线条放松,皱纹抚平改善松弛。可用于化妆品内,作为抗皱成分,且效果极佳。产品参数----乙酰基六肽-8/阿基瑞林中文名称:乙酰基六肽-8/阿基瑞林/六胜肽/乙酰六胜肽-3英文名称:Acetyl Hexapeptide-8/Argireline/Acetyl Hexapeptide-3, CAS号:616204-22-9纯度:≥99%分子量 :888.91g/mol分子式 :C34H60N14O12S外观:白色粉末或液体储存条件:2 ℃~8 ℃包装规格(粉末):1g, 10g, 100g包装规格(液体):20ml/瓶,1KG/瓶应用:化妆品原料功效与应用----乙酰基六肽-8/阿基瑞林抗皱抗衰老改善皮肤质量脸部、颈部和手护理品可添加到美容护肤品中,如乳液、面膜、早晚霜、眼部精华液等作用机理----乙酰基六肽-8/阿基瑞林乙酰基六肽-8参与竞争 SNAP - 25 在融泡复合体的位点, 从而影响复合体的形成。当融泡复合体稍有不稳定, 囊泡不能有效释放神经递质, 从而致使肌肉收缩减弱,防止皱纹的形成。[img=,690,143]https://ng1.17img.cn/bbsfiles/images/2020/10/202010141430498557_1196_3531468_3.jpg!w690x143.jpg[/img]国肽生物主要提供:多肽合成、多肽定制、同位素标记肽、人工胰岛素、磷酸肽、生物素标记肽、荧光标记肽(Cy3、Cy5、Fitc、AMC等)、目录肽、偶联蛋白(KLH、BSA、OVA等)、美容肽、化妆品肽、多肽文库构建、抗体服务、糖肽、订书肽、药物肽、RGD环肽等。详情请咨询国肽生物
关于乙酰氯的国家标准在网上找了很久,找到了份氯乙酰氯的然后发现了乙酰氯GOST 5829-1971乙酰氯 技术条件 (俄罗斯标准)GJB 702-1989 氯乙酰氯 (中国国家军用标准)哪位老师有做过,或者了解乙酰氯国家标准的可否帮下忙,传一份给我,急用,先谢谢大家了
各位老师,乙酰乙酸乙酯说有烯醇类和酮类平衡的混合物。那是不是就是说,我进一针乙酰乙酸乙酯的分析纯,应该出两个峰,一个乙酰乙酸乙酯(烯醇类),一个是乙酯乙酯乙酯(酮类)。
乙酰二茂铁的合成目的原理实验目的 1 通过乙酰二茂铁的制备,了解用Friendel-Crafts酰基化反应制备非苯芳酮的原理和方法。2 学习柱色谱分离提纯产品和薄层色谱跟踪反应进程的原理和操作方法。实验原理 二茂铁又名双环戊二烯基铁,是由2个环戊二烯负离子和一个二价铁离子键合而成。一般认为,以乙酸酐为酰化剂,三氟化硼,氢氟酸,磷酸为催化剂,主要生成一元取代物;如用无水三氯化铝为催化剂,酰氯或酸酐为酰化剂,当酰化剂与二茂铁的摩尔比为2∶1时,反应产物以1,1′-二元取代物为主。二茂铁及其衍生物的分离最好是用层析法。本实验用柱色谱分离提纯产品,可用薄层色谱法跟踪反应进程,柱色谱和薄层色谱均属于吸附色谱,柱色谱分离提纯是根据二茂铁,乙酰二茂铁和1,1′-二乙酰基二茂铁对活性氧化铝吸附能力的差异而进行分离提纯。用薄层色谱跟踪反应进程,根据二茂铁和乙酰二茂铁的斑点大小可以了解乙酰化反应的进程。仪器药品 5ml圆底烧瓶,克莱森接头,干燥管,电磁加热搅拌器,30cm色谱柱(自制),30×100mm载玻片,离心试管50ml烧杯,玻璃钉漏斗,吸滤瓶,锥形瓶,氮气袋,250ml烧杯二茂铁,乙酸酐,85%H3PO4,25%NaOH,二氯甲烷,棉花,洗净的砂,Ⅲ级活性氧化铝,己烷,醇,硅胶,0.5%羚甲基纤维素,干燥氮气。过程步骤 一、乙酰二茂铁的制备称取100mg(0.54mmol)二茂铁,放入5ml圆底烧瓶中,加入2.0ml醋酸酐。装上带有干燥管的克莱森接头。水浴温热并搅拌使二茂铁溶解。移去水浴,打开塞子迅速加入3ml 85% H3PO4,使反应液变成深红色,室温下搅拌1.5h,在反应期间定期用毛细管在液面上吸取2滴左右反应液放入具塞小试管中,假如10滴二氯甲烷,所得溶液用薄层色谱法展开,以了解反应进程。当二茂铁的斑点很浅时,表示反应基本完成。将反应液滴入盛有1g碎冰5ml烧杯中,滴加25%NaOH中和恰至碱性,得到大量桔黄色沉淀。充分冷却后抽滤,1ml冷水分几次洗涤沉淀,抽干,干燥后称重约110~120mg。二、乙酰基二茂铁的柱色谱法分离(1)干法装柱将粗产品溶于0.5ml二氯甲烷加入300mgⅢ级活性氧化铝,振荡均匀得浆状物。在通风橱中,在干燥氮气下除去溶剂至恒重,得到松散的颗粒状物,精确称取1/2用作柱色谱分离。将自制的1.5×30cm色谱柱洗净,干燥,柱底铺一层玻璃棉或脱脂棉,再铺一层约5~8mm厚的砂,填平。称取5gⅢ级活性的中性氧化铝(60~80目),通过漏斗将氧化铝装入柱管内,轻敲柱管,使之填均匀。将精确称得含有1/2产品重的氧化铝装入柱内,顶部盖一层约5mm厚的砂子,使氧化铝顶端和砂子上层保持水平。(2)洗脱用己烷作洗脱剂从柱顶加入,缓慢滴入己烷逐渐展开得到黄色、橙色分离的色谱带。黄色的二茂铁带首先从柱下流出,用己称重的锥形瓶收集洗脱溶液。当黄色谱带完全洗脱下来时,改用体积比为1∶1的二氯甲烷己烷混合物洗脱,同时橙色带往下移动,逐渐改变溶剂的比例到体积比9∶1二氯甲烷己烷混合溶剂时,则将橙色色谱带完全洗脱下来,用另一只已称重的锥形瓶收集洗脱液。最后改用体积比为9∶1二氯甲烷甲醇洗脱时,可以看到很淡的,很少量的,棕色色带向下移动,将该洗脱液另行收集。(3)收集产品在通风橱内,各组分洗脱液分别在水浴上蒸馏,回收溶剂。浓缩后的溶液放置冷却析出结晶,将产品放在盛有石蜡片的干燥器内至恒重。可回收到未反应的二茂铁20~22mg;得到乙酰二茂铁80~90mg 1,1′-二乙酰基二茂铁少于2mg。分别测定熔点。注意事项1.二茂铁需经升华或用石油醚(30~60℃)重结晶纯化。2.仪器应是充分干燥的。3.乙酸酐是临用前经重新蒸馏的。4.吸附剂的活性与其含水量的关,含水量越低,活性越高。氧化铝放入高温炉中(300~400℃)烘3h得无水物即Ⅰ级氧化铝。Ⅲ级氧化铝可用Ⅰ级活性氧化铝加入重量的6%的水而得到。如所用氧化铝活性过强会使产品不易洗脱,浪费较多的溶剂。5.这里是考虑到柱色谱的容器。一般粗产品重75mg以上都仅取1/2作柱色谱分离。6.二茂铁易升华,故测熔点时要封管。熔点的文献值:二茂铁为173℃,乙酰二茂铁为85℃,1,1ˊ-乙酰基二茂铁为130℃。分析思考1. 二茂铁乙酰化反应的机理怎样?2. 怎样利用薄层层析判断乙酰化反应的进程?3. 乙酰二茂铁在石油醚和乙醚中溶解度哪个更大?为什么?4. 柱层析分离二茂铁衍生物时,如何选择展开的溶剂? [img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705162025_52002_1632583_3.gif[/img][img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705162025_52003_1632583_3.gif[/img]
最近同事在做一项目原料是乙酰乙酸甲酯在用原料进行纯度分析的时候,出了很多的峰,而且主峰有拖尾现象色谱条件:检测波长254,流动相:乙腈:水=1:1,流速1.0不知大家用液相做过这产品没有?用的是什么方法?以上的方法该怎么改进?谢谢![em0909]
想请教一下大家,对乙酰氨基酚的紫外吸收波长是多少呀,我查了文献有个说在205nm到230nm之间,但也有用270nm的,我看药典上“有关物质”的测定下用的是254nm,“含量测定”项下用的是257nm,但我在做实验时在254nm波长处检测不到对乙酰氨基酚,不知道是怎么回事,郁闷中……说明:我做的是制备液相,走空白能检测到信号,加对乙酰氨基酚样品后没什么反应,看是以为是浓度不够,后来加大浓度还是检测不到,又以为是254nm波长处没有吸收,但药典也是254nm呀,检查了可能出现的问题,也没找到为什么, 为什么空白都能检测到信号,进样后检测不到呢???很郁闷 请大家指教!!
想请教一下大家,对乙酰氨基酚的紫外吸收波长是多少呀,我查了文献有个说在205nm到230nm之间,但也有用270nm的,我看药典上“有关物质”的测定下用的是254nm,“含量测定”项下用的是257nm,但我在做实验时在254nm波长处检测不到对乙酰氨基酚,不知道是怎么回事,郁闷中……说明:我做的是制备液相,走空白能检测到信号,加对乙酰氨基酚样品后没什么反应,看是以为是浓度不够,后来加大浓度还是检测不到,又以为是254nm波长处没有吸收,但药典也是254nm呀,检查了可能出现的问题,也没找到为什么, 为什么空白都能检测到信号,进样后检测不到呢???很郁闷 请大家指教!!
求助,在使用乙酰丙酮方法测甲醛时,为何配制好的乙酰丙酮溶液颜色很黄?注:所用的乙酰丙酮原本无色,但乙酸铵有点回潮。
利用waters液相分析甲胺磷和乙酰甲胺磷时,色谱柱为反相C18,流动性为水和乙腈。但通过改变流动相比例(水:乙腈=97:3,90:10,80:20,梯度:99:1 1min,98:2,97:3),改变流速(1,0.5),柱温(30,35 ,40),两种农药一直分不开。求助各位大神帮忙,急!急!!!!
最近做绿茶提取物甲胺磷,乙酰甲胺磷,对硫磷的检测,回收率做不出,甲胺磷乙酰甲胺磷才4-5%,对硫磷有70-80%。我使用的是安捷伦的6890N,检测器NPD,不分流进样,进标准品1.0ug/ml峰面积大概是300左右,但是不是很稳定。感觉问题出在前处理,分别用正己烷,乙酸乙酯,乙腈做过过加样回收,加2ml浓度1.0ug/ml。乙酸乙酯,乙腈提取浓缩后为粘稠红棕色液体,用无水硫酸镁和活性炭分散固相萃取后没有改善,正己烷提取后浓缩液无色。浓缩前进过仪器是可以测出来的,浓缩后没有了,回收液液没有,估计分解了。浓缩使用的是步其的平行定量浓缩仪,水浴45度,真空度250Mbar,冷凝水10-13度。我认为是浓缩步骤出的问题,大家帮分析一下,到底是哪里出的问题?有没有跟好的前处理分享一下,要方法简单的哦
我们想用GC来检测乙酰氧基乙酰氯,因为原来是采用滴定方式,过程比较烦琐,而且结果准确度不高。
现我厂采购两种乙酰氯,一种是有磷的,一种是无磷的,用酯化法检测后有磷乙酰氯与生产厂家结果分析误差比较大,什么原因?各位谁知道
我的是Agilent 7809A,配FPD檢測器,使用DB-1701,30mx0.53mmx0.25um的色譜柱,進樣濃度分別是500ng/ml的甲胺磷及乙酰甲胺磷,響應值分別是8000及5000,而且峰型很好,但是當我轉用HP-5,30mx0.32mmx0.25um色譜柱時,進同樣的濃度,所有條件不變,只是把柱流量調低至1ml/min,甲胺磷及乙酰甲胺磷只是出現微微的駝峰,安全不成峰,同時發現粗直徑較幼直徑的色譜柱適合檢測甲胺磷及乙酰甲胺磷,大家是否有同感,還是我有地方混淆了,大家討論一下
我要做农药残留检测,找不到乙酰胆碱酯酶,可直接用酶片(京蓬生物科研有限公司产的)代替吗?[em63]
不知哪位大哥有乙酰羟胺的分析方法?多谢!
我们实验室最近用乙酰丙酮法测定空气中的甲醛,发现标准系列灵敏度不如酚试剂法。请问大家用乙酰丙酮法测过甲醛没有,测的话,一般标准系列吸光度应该是多少呢???可否发出来做一个参考?非常感谢!!!
乙酰甲胺磷的问题我直接进样一定系列浓度的乙酰甲胺磷,线性很差,只有0.992,但是进样其他的,譬如敌敌畏,其他的线性都很好,不知道为什么
大家好,我现在在做乙酰乙酸甲酯气相色谱检测,用同一根色谱柱进样,有时候时两个峰,有时候是一个峰到底是什么原因?乙酰乙酸甲酯有互变异构体,在运行过程中有出现两个峰的可能。 现在能找到乙酰乙酸甲酯和乙酰乙酸乙酯的国标。乙酰乙酸甲酯的国标中是一各峰,而乙酰乙酸乙酯是两个峰,这两个东西的性质是相似的,为什么会出现不同的峰形呢?
氯乙酰氯[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析时氯乙酸、三氯甲苯、苯甲酰氯、苯甲酸几种组分的含量重复性很差,是什么原因?哪位老师有氯乙酰氯比较简便实用的分析方法?







 我要推广仪器
我要推广仪器
 下载APP
下载APP