吡啶二羧酸酐结构看似简单,可因其在水中水解成酸,影响样品游离酸含量分析,化学滴定不好作;做GC时热分解,与相应的二酸峰重合;液相用水溶液作流动相不行,用非水有机相作流动相如用醇又会醇解,估计可用正相做,但该样在许多有机溶剂中溶解性差,所以一直没找到好的分析方法。请问哪位老师作过吡啶二羧酸酐的HPLC分析,能否帮帮我。谢放!
一个实例,用七氟丁酸酐可以与二苯基甲烷二胺发生衍生化反应,衍生化的机理应该在于胺根上面。但二苯基甲烷二胺里面的胺还会与羧酸发生反应。如果同时向含有 大量的七氟丁酸酐 和 极少量的 丁酸 的混合溶剂中,加入少量二苯基甲烷二胺,请问二苯基甲烷二胺是先发生衍生化反应还是与丁酸反应?
据说欧盟委员会在官方公报上发布决议(EU) 2018/594,正式将苯-1,2,4-三羧酸-1,2-酐(偏苯三酸酐)(TMA)确定为高度关注物质(SVHCs)。而在官网链接中还是181项[url]https://echa.europa.eu/web/guest/candidate-list-table[/url];是不是还没加进清单呢?[img]http://simg.instrument.com.cn/bbs/images/default/emyc1010.gif[/img]
[color=#444444]请问各位大侠,羧酸和羧酸盐在同一液相色谱条件下出峰的时间是否一样?例如醋酸和醋酸钠,流动相为乙腈和水,PH为酸性,磷酸做缓冲液。另外,羧酸和羧酸盐的极性是否相同?[/color]
各位大神,我要测一个原料药里的丁二酸酐,但是丁二酸酐性质不稳,遇水会水解,即使是流动相里有水,也会水解,请问如何用LC-MSMS对丁二酸酐进行定性定量?
我今天用乙酸酐衍生处理氯酚类,结果都没出峰。实验室买的乙酸酐放置的时间久了,将乙酸酐加到碳酸钾溶液,加进去的时候都是像一个圆球一样沉在碳酸钾溶液里面,类似于将不溶于水的有机溶剂滴加到水里面。但是振荡之后又逐渐溶于碳酸钾溶液了。是不是乙酸酐变质失效了?乙酸酐变质后应该是乙酸,乙酸应该易溶于碳酸钾溶液啊。[img]文件不能大于5M[/img]
请问同志们,羧酸类物质能检测吗??能用甲醇做流动相吗?羧酸会不会和甲醇生成酯,而影响测定羧酸的PH=3-4,需要配制流动相+怎么样的缓冲溶液??????大家帮忙啊
做实验要用到苯二甲酸酐,但是都买不到,都是只有邻苯二甲酸酐,问了几家试剂公司,有的说是一个东西,有的说不是,我在网上查苯二甲酸酐,出来的也都是邻苯二甲酸酐,到现在也没有确切的答案,但是国标方法上就写的苯二甲酸酐.
[color=#444444]3-异丁基戊二酸酐,bp=279℃,可能含有醋酸酐[/color][color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的检测纯度条件是?具体点儿,温度[/color][color=#444444]谢谢[/color]
求用羧酸合成酰 氯,羧酸分子350以上,酰 氯如何分析,酰 氯反应如何中控?
本人试验所得的糖类水溶液,欲先旋蒸将水除去,然后用醋酸酐跟吡啶混合溶液乙酰化,有几个问题想向大家请教:1.,醋酸酐与吡啶的比例是多少?2,乙酰化的温度是多少?3,反应后的吡啶以及过量的醋酸酐怎么除去?
本人试验所得的糖类水溶液,欲先旋蒸将水除去,然后用醋酸酐跟吡啶混合溶液乙酰化,有几个问题想向大家请教:1.,醋酸酐与吡啶的比例是多少?2,乙酰化的温度是多少?3,反应后的吡啶以及过量的醋酸酐怎么除去?
国标上写的 苯二甲酸酐C6H40 邻苯二甲酸酐的化学式是 C8H4O3 是怎么回事呢,着急呀
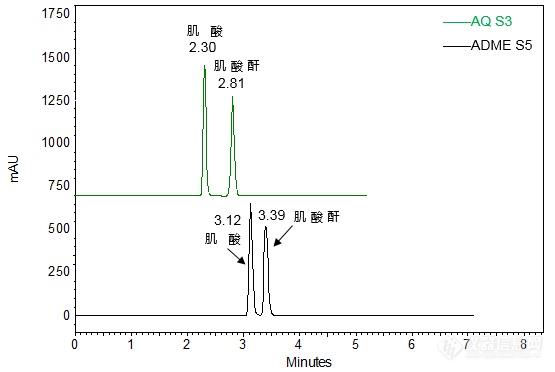
[align=center][b]肌酸和肌酸酐的分析[/b][/align][b][/b]客户提供肌酸、肌酸酐样品,希望实验室帮忙实现二者的良好分离与保留。由于肌酸和肌酸酐极性较强,我们首先尝试在酸性条件下(0.05%磷酸),使用高表面极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ S3与CAPCELL PAK ADME S5两款色谱柱,分别对二者混合样品进行分析,结果如图1所示,肌酸的保留时间分别为2.30 和3.12 min,保留较弱。[align=center][img=,555,372]http://ng1.17img.cn/bbsfiles/images/2017/11/201711021020_01_2222981_3.png!w555x372.jpg[/img][/align][align=center]图1 肌酸、肌酸酐混合溶液分析色谱图(0.05%磷酸溶液)[/align]*注:峰上标数字为保留时间。[img=,419,152]http://ng1.17img.cn/bbsfiles/images/2017/11/201711021020_02_2222981_3.png!w419x152.jpg[/img]为增强保留,尝试在中性条件下,以200 mmol/L高氯酸钠溶液为流动相对肌酸和肌酸酐进行分析,结果如图2所示。使用AQ S3色谱柱进行分析时,肌酸、肌酸酐的保留时间分别为3.36 min、5.23min。使用ADME S5色谱柱进行分析时,肌酸、肌酸酐的保留时间分别为3.48 min、6.29 min。ADME 色谱柱较AQ S3 色谱柱整体保留较强。[align=center][img=,550,378]http://ng1.17img.cn/bbsfiles/images/2017/11/201711021022_01_2222981_3.png!w550x378.jpg[/img][/align][align=center]图2 肌酸、肌酸酐混合溶液分析色谱图(200mmol/L高氯酸钠溶液)[/align]*注:峰上标数字为保留时间。[img=,420,149]http://ng1.17img.cn/bbsfiles/images/2017/11/201711021022_02_2222981_3.png!w420x149.jpg[/img]进一步将200mmol/L高氯酸钠溶液的pH调至酸性(pH=2.74),以ADME S5色谱柱对肌酸、肌酸酐混合样品进行分析,结果如图3所示。肌酸、肌酸酐保留时间分别延长至4.45、5.07,二者间分离度为3.12,达到基线分离。[align=center][img=,552,372]http://ng1.17img.cn/bbsfiles/images/2017/11/201711021023_01_2222981_3.png!w552x372.jpg[/img][/align][align=center]图3 肌酸、肌酸酐混合溶液分析色谱图(200 mmol/L高氯酸钠溶液、pH 2.74)[/align]*注:峰上标数字由下至上依次为分离度与保留时间。[img=,407,138]http://ng1.17img.cn/bbsfiles/images/2017/11/201711021023_02_2222981_3.png!w407x138.jpg[/img]
HJ 1220-2021 6种羧酸类的测定 [url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]质谱法 一般的柱子的可以分析吗
根据GBZ/T 160.67-2004 扩项工作场所MDI。原理:空气中MDI用冲击式吸收管采集,水解后成4.4’-二氨基二苯甲烷(MDA),在碱性条件下用甲苯萃取,经七氟丁酸酐衍生后,取甲苯溶液进样,经色谱柱分离,电子捕获检测器检测,以保留时间定性,峰面积定量。标准曲线的绘制:在5 只干燥的具塞离心管中,0.0、0.25、0.50、1.0和2.0ml MDA标准溶液,用甲苯稀释至2.0ml,配制成0.0、0.025、0.050、0.10和0.20mg/ml MDA标准系列,各管加30ul 七氟丁酸酐,振摇2min,放置5min,加1ml 缓冲液,振摇2min,以除去过剩的七氟丁酸酐,放置2min,将甲苯层转移入另一离心管中,供测定。色谱柱 DB-5 柱温230℃ 进样器270℃,检测器250℃,结果只出甲苯溶剂峰。参考了些文献七氟丁酸酐与胺反应有加热55℃70分钟的,也有反应30min的。标准里衍生反应很短时间怀疑衍生反应有问题!
我看的文献方法衍生全氟羧酸,用三乙基硅烷醇的方法,用的仪器是岛津的单杆EI 源,但是衍生以后全扫模式下,所有的全氟羧酸出的峰都一样。通过SIM模式下才能找到目标峰,并且PFDA/PFNA/PFDOA的峰都非常小。我用的是1ug/ml得标液衍生的,全氟辛酸的峰大概只有1000,其他的峰高就只有100不到。有没有大神做过类似的方面,求帮助。还有一个问题,如果做全氟羧酸的目标物,用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]做的话,文献中有用NCI源和EI源的,具体的那个方法更好一点呢。跪谢!
请教: 甲基磺酸酐的纯度分析方法? 请多多指点!甲基磺酸酐Methanesulfonic anhydrideDensity:1.583 g/cm3Melting Point:64-67 °C(lit.)Boiling Point:289.3 °C at 760 mmHgFlash Point:128.7 °CAppearance:white to light brown to grey powder, crystals
工作场所羧酸类,我只有ffap极性毛细管柱,方法中乙酸是用甲酸作为溶剂,可是甲酸腐蚀性太大,不敢做,有没有其它溶剂能代替的,还有氯乙酸响应实在太小,峰型很难看,检出限远达不到国标,是色谱柱的问题吗?哪位同行做过,详解一下
请问:羧酸类打[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]峰型总是不好,怎么办?
请各位高手指教下醋酸酐的检验方法,用气质联用进了个标准,进了样品,出峰 的时间基本一致,因为库里面没有醋酸酐,不能定,求大家指教一个检验醋酸酐的方法。
液相色谱怎么测酸酐?
做金属物质检测时,会用的吡啶-2,6-二羧酸,皮考啉二酸,DPA,这个物质,不知道有没有那个老师以前做过?实验过程中,起到什么作用呢?谢谢!
羧酸键合硅胶柱用前需要怎么处理,用后如何保养,求指教
Porapak T能出醋酸酐峰吗?分析醋酸酐反应液用什么填充柱?
比如,丁酸酐或者丙酸酐,溶剂该怎么选?柱子?分析过该类物质的版友分享一下经验。
如何将样品中醋酸和醋酸酐的峰分开
请问一下,工业用马来酸酐,想要检测其纯度,有没有国家标准的?
有朋友做过氯桥酸酐GC分析吗?请问用什么GC柱,柱温、汽化和检测温度分别是多少?谢谢!
买了一根羧酸键合硅胶柱要保存在叠氮钠里边,但采购说没有优级纯只有分析纯,可以吗



 我要推广仪器
我要推广仪器
 下载APP
下载APP