DNP混合柱,原来一直使用正常,最近突然出现内标与异戊醇无法分离的现象,可是其他的峰都正常,不知道什么原因?
什么样的填充柱对甲醇吸附较大,苯分离效果显著 我想分析甲醇中的苯系物,甲醇拖尾太大,无法精确定量.请教高人指导.
用国家标准做水产品中的组胺,正戊醇提取时无法分离有机相,why?有何窍门?

最近在做仪器检定,流动相为85%甲醇水溶液,激发波长:290nm,发射波长330nm,标品为1x10-4 g/mL的萘-甲醇溶液。因为才接触液相,之前做的样出的峰都是比较标准的,这次萘甲醇出来的峰没有尖,后来发现所有重复的12个样都是这样的峰形(见下图)。但是在输入时间后不能显示保留时间,无法积分,看不到峰面积和保留时间,很是困惑,不知道大家有没有人在用荧光检测器在做萘甲醇?可不可以发一下大家做的图谱呢?http://ng1.17img.cn/bbsfiles/images/2016/12/201612281211_01_3167224_3.jpg
我用岛津GC2010Plus,WAX色谱柱测定甲醇中的苯系物,其中苯峰与甲醇试剂峰无法分离,请问各位大虾如何解决这个问题?
用FFAP分析白酒中乙酸乙酯和甲醇时,乙酸乙酯和甲醇的出峰时间比较接近,如果乙酸乙酯含量太高,甲醇含量太低的时候,乙酸乙酯出峰覆盖甲醇峰,无法对甲醇的浓度做出判断!请问该用什么方法解决?
甲醇是不是voc挥发性有机物
国产新药和进口药物的价格往往相差几倍,一些用于治疗慢性病或者癌症的生物制药,价格更要相差10倍甚至几十倍。其中的关键问题,不是进口药物的成分更新,而是他们的提纯工艺比国内更成熟,这使得进口药物的副作用更小。不少生物制药行业以及化工行业的专家学者提出,必须加快生物制药行业的系统发展,尤其要加快提高化学行业的提纯技术,否则,即使研究人员对药物的机理和靶点了解得再清楚,也没有办法制造出高质量的药品。据有关专家介绍,制药业是化学原料的分解、合成技术与现代临床诊断医学相结合的制造工业,也是衡量一个国家制药能力和水平的主要标志之一。我国目前制药市场的增幅每年在15%左右,5年后,有望成为全球第七大制药市场。但是我国的制药行业面临诸多困难,尤其是随着现代生物医药的发展,现在很多药物都强调靶点越单一越好,即药物成分只单纯针对某一类疾病。而我国有限的提纯工艺水平,导致药物单体成分纯度不够高,进而使药品失去了竞争力。专家告诉记者,一种药物中有很多种成分,每个成分都有自己的分子结构,即所谓单体。现在制药界更倾向于制造单体纯度高并且成分单一的药物,因为成分单一可以使药物的作用更加明显,副作用更少。比如针对β受体的药物,如果针对心血管β受体,所起的作用应该是降压;如果是呼吸道的β受体,针对的疾病应该是哮喘。但是如果这些药物中的成分还同时针对胰腺β受体,那么对病人来说,就可能是一场灾难。有些治疗高血压等疾病的药物会引起血糖高甚至导致糖尿病,就是因为药物中含有的β受体抑止剂单纯性不够。而大多数药物如果要实现成分单一的话,就必须从化合物中或者是植物中提纯。据了解,药物提纯工艺不够先进,一直是制约我国生物制药发展的一个瓶颈。因为生物制药最重要的环节,就是将有效成分从蛋白质中分离出来。华东理工大学制药专业的一位教授称,如果是在实验室环境中,不惜一切代价地从一种植物中或者是化合物中提纯出某种单体药物成分,这并不难,但是要作为产品大量生产,我国不论从生产工艺上还是从化学提纯的角度,都无法做到。目前化学提纯工艺在国外已经受到越来越多关注,但是我国制药企业在这方面的研发投入仍然很低。专家介绍说,中国制药业的研发更多地重视筛药,常常是一个成分不行就换一种成分,即使是生产工艺方面的研发,也只是着眼于工艺路线的打通,而忽视了工艺过程的优化,尤其是降低副产品的含量等。有关专家认为,如果仅仅能够完成成分筛选,发现针对某种疾病的有效成分,而无法将它从植物等中提纯出来的话,这些成果仍然无法应用在临床上。有关专家呼吁,加速生物医药发展是一项系统工程,不能仅仅关注生物医药本身,而应该加强相关行业每个环节的发展。
请问各位有没有人做过北京坛墨的甲醇中的五种挥发性卤代烃。学习过程中,请各位老师帮帮忙!
这两天小朋友做白酒中的氰化物,觉得试剂空白也显色。于是,我让她把所有试剂都重新配了,再做,还是显色。小姑娘也很认真,一个一个试剂测试过去。(最早我们就排除了容器不干净),然后重新拿了容器临时再配试剂。最后发现,是用作指示剂的酚酞溶液出现了氰化物问题。酚酞溶液的溶剂是用95%乙醇配制的。用作酒类碱解后,中和后的pH调整指示,一般就加一两滴。我们用默克的氰化物快速检测盒迅速的测试了下其含量, 颜色很深,时间未到就已经超出标曲的最高点了。也就是为什么我们只加一两滴用来调试pH值,试剂空白都能显色(已经明显干扰到我们的实际检测了)。这个是国药沪试的95%乙醇,然后我们又测试了同家的无水乙醇,同样很高。 然后还测试了山东禹王的色谱级的应该是无水乙醇吧。同样也有氰化物。再翻出了一两年前生产的国药的无水乙醇,干干净净。-----------出现这个问题后,我们看了国药沪试的质检报告,里面没有氰化物,所以也没有办法去要求什么, 但也如实的联系了其质检部,反应了情况。因为含量很高,所以我还是认为这个是不正常的。所以就算没有办法要求什么,但能反应的还是反应,如果厂家能重视,应该能提高试剂的品质。-----------刚才有人私下里问说,具体数值。因为我们没空具体测,所以用快速测试盒蛮先看了下。 最高点0.030mg/L,我们取样是标准取样的1/4,那么至少在0.12mg/L以上。
大家好,我们厂因为大量使用液体新戊二醇,浓度要求在90%,水分含量在10%左右,误差不能高于1%,用检测折光率的办法比较迅捷,但准确度不是很高,如果里面掺杂其他液体有机醇,折光率法无法检测出来,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测吧,该物质在60摄氏度以下,容易结晶,请问各位高手有什么好的办法?谢谢!!!
请问谁有环保部标样所生产的甲醇中10种挥发性有机物(I)的标值啊,救急救急!!!
相比于京霾的沉重,晋霾的激烈,沪霾的湿热和粤霾的阴冷,我更喜欢冀霾的醇厚,它是如此的真实,又是如此的具体。黄土的甜腥与秸秆焚烧的碳香充分混合,再加上尾气的催化和低气压的衬托,最后再经热源袅袅硫烟的勾兑,使得冀霾特别是廊坊霾口感干冽适口,吸入后挂肺持久绵长,让品味者肺腑欲焚,欲罢不能。这是人类辛劳与自然的馈赠共同作用的结晶。雾是帝都重,霾是故乡醇! 不说了,眼快熏瞎了!http://simg.instrument.com.cn/bbs/images/default/em09510.gif 你家乡的雾霾肿么样了?上传个照片让大家瞧瞧?顺便对比对比呀!
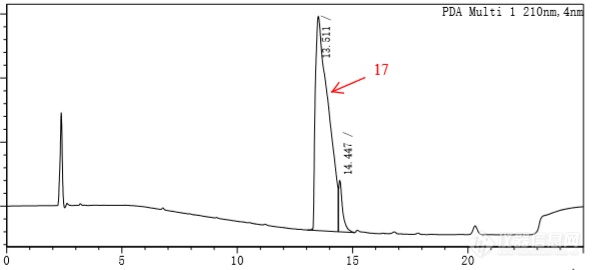
这个化合物含有醛基,多手性中性,可能有多个异构体,分子量为790.94,极性很弱。用以前的方法,50%乙腈和50%0.1%TFA为起始比例,走梯度,峰底很宽,多个化合物分离不开,全部包含在内。图如下。我后面又尝试了几种方法,都分不开。[img=,593,270]https://ng1.17img.cn/bbsfiles/images/2019/05/201905091224341957_1125_3116636_3.png!w593x270.jpg[/img]尝试方法一:在乙腈中加30%异丙醇,其余不变,出峰情况如下:[img=,592,274]https://ng1.17img.cn/bbsfiles/images/2019/05/201905091227411711_5958_3116636_3.png!w592x274.jpg[/img]尝试方法二:增加异丙醇比例到40%[img=,591,260]https://ng1.17img.cn/bbsfiles/images/2019/05/201905091229177104_356_3116636_3.png!w591x260.jpg[/img]尝试方法三:有机相30%异丙醇+70%乙腈,初始比例为25:75,等梯度运行:[img=,623,277]https://ng1.17img.cn/bbsfiles/images/2019/05/201905091234166027_3835_3116636_3.png!w623x277.jpg[/img]尝试方法四:更换C8色谱柱,方法与1相同:[img=,617,271]https://ng1.17img.cn/bbsfiles/images/2019/05/201905091231078275_7956_3116636_3.png!w617x271.jpg[/img]也尝试了其他的,比如C8下更改流动相比例,C18下更改流速。但是依然无法改善分离情况。是否有其他在不换柱子的情况下改善出峰情况的办法?如果换柱子,换什么柱子比较合适?)————————————————————分割线我在里面加入了甲醛后,发现分离效果比较明显,但是有个很大的问题,那就是后面一个峰不见了,怀疑缩合了,醛与甲醇中的醇反应!!如果不加甲醇,还有其他加入的溶剂来改善吗?有没有遇到过的大神推荐一下。
在做液相色谱质谱检测白藜芦醇及其衍生物的实验,但是做标准品的时候就无法分离白藜芦醇和白藜芦醇苷。流动相采用的甲醇和水,等度洗脱和梯度洗脱都试过。色谱柱是安捷伦的C18,内径是2.1mm,膜厚用过3.5和5.0的,柱长从50mm到150mm都试过。现在用的是 Thermo Golden(4.6*250mm,5μm)的柱子,还是无法分离。求各位大神指教!!!
[color=#444444]在做液相色谱质谱检测白藜芦醇及其衍生物的实验,但是做标准品的时候就无法分离白藜芦醇和白藜芦醇苷。流动相采用的甲醇和水,等度洗脱和梯度洗脱都试过。色谱柱是安捷伦的C18,内径是2.1mm,膜厚用过3.5和5.0的,柱长从50mm到150mm都试过。现在用的是 Thermo Golden(4.6*250mm,5μm)的柱子,还是无法分离。求各位大神指教!!![/color]
我有一硫醇类化合物,液体,用普通卡尔费休试剂无法测定,求助一下,目前用什么样的方法可以测定?在网上有一篇缓冲盐配制测定的方法没有太大效果。
我单位PE2100DV ICP 点火后总提示雾化气流无法维持,雾化器清洗了n遍、两个宝石喷嘴也更换了新的,检查红宝石气源的电磁阀也带电,阀也是好的。工作时雾化器检测流量为0.8,成分能正常检测。但为啥总提示雾化器气流无法维持?

求助各位大神帮忙看看,热解析法做甲醇中苯系物,谱图这样可以吗?苯峰还是拖在甲醇峰上[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/03/201903261617262266_5419_3497108_3.png[/img]
各位,GC做有机氯时,发现:标物为正己烷基体中9种有机氯(666、DDT、六氯苯),分别以正己烷和甲醇为基体稀释20倍,且分别向两种基体中加同量微量丙酮(丙酮为助溶正己烷/甲醇体系,为使两种体系具有可比性,所向两种稀释的标准中都加了同量微量丙酮),发现正己烷基体中9种出峰情况正常,但甲醇基体中γ-666,δ-666,op-ddt,pp'-ddt这四种物质出峰很低(2种DDT可明显判断是分解了,但γ-666和δ-666无法有效确证,因为666是有大π键是很稳定的),请各位大侠指教。(已排除衬管惰性降低的可能,这一点请各位不用考虑,而且多次实验表明:同样浓度,甲醇基体的有机氯标物中以上4种物质峰高明显比正己烷基体的有机氯标物要低很多)多谢!见附件中谱图,图中画圈的为9种目标组分,第三个较高的峰为六氯苯。欢迎可加QQ289638727沟通!
在技术版面里发贴时图片无法上传,上午就是这情况了,下午还是如此,怎么回事?
挥发性有机物测定,甲醇中苯系物都是7种物质,没有异丙苯,只有二硫化碳种苯系物有8种的标准溶液,大家都买的甲醇还是二硫化碳的呢?
化合物纯度的鉴定方法,从快速,便宜,简便的要求出发,主要来之于以下几点:一 通过TLC的纯度的鉴定, 我将自己的心得分述如下1 展开溶剂的选择,不只是至少需要3种不同极性展开系统展开,我的经验是首先要选择三种分子间作用力不同的溶剂系统,如氯仿\甲醇,环己烷\乙酸乙酯,正丁醇\醋酸\水,分别展开来确定组分是否为单一斑点.这样做的好处是很明显的,通过组份间的各种差别将组分分开,有可能几个相似组份在一种溶剂系统中是单一斑点,因为该溶剂系统与这几个组分的分子间力作用无显著的差别,不足以在TLC区分.而换了分子间作用力不同的另一溶剂系统,就有可能分开.这是用3种不同极性展开系统展开所不能达到的.2 对于一种溶剂系统正如wxw0825所言,至少需要3种不同极性展开系统展开,一种极性的展开系统将目标组分的Rf推至0.5,另两种极性的展开系统将目标组分的Rf推至0.8,0.2。其作用是检查有没有极性比目标组分更大或更小的杂质。3 显色方法,光展开是不够的,还要用各种显色方法。一般一定要使用通用型显色剂,如10%硫酸,碘,因为每种显色剂(不论是通用型显色剂,还是专属显色剂在工作中都遇到他们都有一化合物不显色的时候),再根据组分可能含有混杂组份的情况,选用专属显色剂。只有在多个显色剂下均为单一斑点,这时才能下结论样品为薄层纯二 通过熔程,判断纯度。原理很简单,纯化合物,熔程很短,1,2度。混合物熔点下降,熔程变长。三,基于HPLC的纯度鉴定,对于HPLC因为常用的系统较少,加之其分离效果好,我们一般不要求选择三种分子间作用力不同的溶剂系统,只要求选这三种不同极性的溶剂系统,使目标峰在不同的保留时间出峰。四,基于软电离质谱的纯度鉴定。如ESI-MS,APCI-MS。大极性化合物选用ESI-MS,极性很小的化合物选用APCI-MS,这些软电离质谱的特点是只给出化合物的准分子离子峰,通过正负离子的相互沟通来确定分子量。如果样品不纯,就会检出多对准分子离子峰,不但确定了纯度,还能明确混杂物的分子量。五,基于核磁共振的纯度鉴定,从氢谱中如果发现有很多积分不到一的小峰,就有可能是样品是样品中的杂质。利用门控去偶的技术通过对碳谱的定量也能实现纯度鉴定。好了,不能再多写了。这里只是对常见的纯度鉴定方法做了一个小结,从快速,便宜,简便的要求出发,以第一点最合要求,往后次之,所以对第一点详加讲述。当然每种方法多有各自的局限性,如基于氢谱的纯度鉴定,如果发现有很多积分不到一的小峰,还有可能使样品中的活泼质子,基于软电离质谱的纯度鉴定,如果混杂物的分子量与目标物一样就无法检出。等等还有很多。这需要大家在工做中积累,思考。要讲的话,我看好几篇都讲不完。最后说一下对化合物纯度的要求,世界上不存在100%纯的化合物。你希望要多高的纯度应该与你的目的有关,例如,如想测核磁共振鉴定结构,一般要求95%的纯度,如果想测EI-MS,纯度越高越好。99%以上。还有,以上的方法都不能区分对应异构体。
新华社东京1月19日电 (记者蓝建中)肠出血性大肠杆菌导致食物中毒时,重症患者有时会出现痉挛和昏睡等脑部症状,甚至会死亡。东京大学和富山大学的研究小组发现,如果在短时间内给重症患者大量注射类固醇,可能会取得较好效果。 2011年,日本烤肉连锁店“惠比寿烤肉酒家”的生拌牛肉引发集体食物中毒事件,导致5人死亡,食物中毒是由大肠杆菌O111引发的。 研究小组对富山、石川和福井等地的86名患者的治疗记录进行了分析。在这些患者中,有34人出现了伴随着肾功能衰竭的溶血尿毒综合征,有21人出现了痉挛和昏睡等脑部症状。 在最初进行治疗时,医生采用的是血浆交换等治疗手段,但是在开始注射类固醇之后,就不再出现死亡病例,在接受类固醇治疗的12名脑部症状患者中,有11人没有出现后遗症而康复。研究小组指出,这参考了治疗流感病毒导致的脑部症状时使用类固醇的做法。 在大肠杆菌O111导致的食物中毒中,通常有很多患者会出现脑部症状。东京大学教授水口雅指出:“对于食物中毒导致的脑部症状,一直缺乏有效的治疗方法,这个发现有可能促进开发出新的治疗方法。” 这一成果论文已经刊登在最新一期美国《神经病学》杂志网络版上。
热解析法测甲醇中苯系物,直接进样的方式进标样出峰正常,通过吸附管进样,三连峰分不开,是什么原因,该怎么解决呢?
热解析法测甲醇中苯系物,直接进样的方式进标样出峰正常,通过吸附管进样,甲苯溶剂峰对苯有干扰,改用二硫化碳也不行,请问是什么原因,该怎么解决呢?
[b]问题:31种挥发性有机物混标,适用于《HJ 642-2013 土壤和沉积物 挥发性有机物的测定 顶空/[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱法》,1000 μg/mL在甲醇中,1 mL/安瓿,货号是?答案:46592=======================================================================【活动内容】1、每个工作日上午10:00左右发布一个色谱问答题,版友根据题目给出自己理解的答案。2、每个工作日下午15:10公布参考答案。【活动奖励】幸运奖:抽奖软件,当天随机抽取3个或5个回答正确的版友ID号(最后一个ID号,截止至下午15:00),每人奖励[color=#ff0000]2钻石币[/color](抽奖人数≤10,抽取3个版友;抽奖人数>10,抽取5个版友);中奖名单:捌道巴拉巴巴巴(注册ID:v3082413)千层峰(注册ID:jxyan)WUYUWUQIU(注册ID:wulin321)zengzhengce163(注册ID:zengzhengce163)zgx3025(注册ID:v2844608)[img=,690,388]http://ng1.17img.cn/bbsfiles/images/2018/06/201806111505464501_1625_1610895_3.jpg!w690x388.jpg[/img][img=,690,388]http://ng1.17img.cn/bbsfiles/images/2018/06/201806111505032361_1723_1610895_3.jpg!w690x388.jpg[/img]积分奖励:所有回答正确的版友奖励[color=#ff0000]10个积分[/color](幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次[/b][align=left][color=#ff0000][b]PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。[/b][/color][/align][align=left][color=#ff0000][b] 下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。[/b][/color][/align][color=#ff0000][b][/b][/color]
乙醇不挥发物,大家做的峰都是怎么样的?帮我分析一下为什么我的甲醇峰跑成这样?用的DB1301的柱子 60m*320μm*1μm 6分多些的是甲醇的峰。
各位前辈好,第一次做SPE,用的是bond elut env(200mg/3ml)的柱子,进行尿液挥发性有机化合物的代谢物的测定,包括巯基尿酸类的代谢物等,物质极性比较大,呈酸性,SPE过程为2ml甲醇,2ml 0.1%甲酸水活化柱子,盐酸调节尿液至ph为2左右上样,以2ml0.1%甲酸水,2ml0.1%甲酸水-甲醇(1:9)淋洗,洗脱用的是1%甲酸甲醇洗脱现在问题是洗脱溶剂似乎不够强,无法将部分待测物洗脱下来,但是淋洗液中却都有百分之10左右的待测物,请问下各位前辈应该如何改善洗脱液
检测血清中的一类化合物,用三氯乙酸沉淀蛋白再用氨水甲醇调至弱酸性,WAX小柱依次用3mL甲醇、3mL水活化。 结果发现在上样的过程中,流出液变成了碱性,把氨水甲醇换成NaOH后也是一样的情况。 用甲醇淋洗的时候,流出液为碱性,但用水淋洗的时候流出液又变成酸性了,用试纸测试甲醇和水均为中性。 不知大家在工作中有没有发现这种情况,出现这种现象的原因是什么?







 我要推广仪器
我要推广仪器
 下载APP
下载APP