求助一下各位老师,用于饲养肉牛的牧草,需要检测农残,应该参照哪个标准来做呢?
NY/T2129-2012牧草取样标准 谢谢
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=101288]NY/T 1574-2007 豆科牧草干草质量分级[/url]
NYT 1692-2009 热带牧草品种资源抗性鉴定柱花草炕炭疽鉴定技术规程[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=174288]NYT 1692-2009 热带牧草品种资源抗性鉴定柱花草炕炭疽鉴定技术规程.pdf[/url]

2010年版药典(一部)中,对益母草中盐酸水苏碱的测定有如下描述(以丙基酰胺键合硅胶为填充剂):http://ng1.17img.cn/bbsfiles/images/2011/01/201101080907_272670_801_3.jpg那么为什么要用丙基酰胺柱来测盐酸水苏碱呢?丙基酰胺硅胶基质的柱子是什么柱子呢? 首先我们要了解盐酸水苏碱的特性,盐酸水苏碱的极性极大,普通的反相色谱对它的保留分离能力较弱,通常在死时间里流出而无法得到分离,而亲水作用色谱HILIC能为极强性的化合物提供良好的保留,在此类化合物上应用广泛。 目前已有多种商品化的HILIC色谱柱,多为硅胶基质,键合不同极性基团,如丙基酰胺基,酰胺基,聚琥珀亚酰胺等极性基团,氨基键合硅胶柱由于使用寿命较短,键合相容易流失,造成保留 丙基酰胺键合硅胶克服了传统正相色谱柱在水相条件下不稳定的缺点,其常使用流动相是和反相色谱相同的水相缓冲液( 40%)及有机溶剂,但是其梯度条件通常是初始为高比例有机相,逐步加大水相含量;极性丙基酰胺键合硅胶的HILIC色谱柱在反相条件下,可以有效的保留极性化合物,是一种崭新的极性化合物HPLC分离解决方式.博纳艾杰尔推出的Venusil HILIC (丙基酰胺键合硅胶),就是一样一款非常适合于益母草中盐酸水苏碱测定的柱子,测定方法及谱图如下:色谱柱:Venusil HILIC (丙基酰胺键合硅胶),4.6×250mm,5µm,100Å(订货号:VH952505-0)流动相:乙腈-0.2%冰醋酸(80:20)流速:0.5mL/min柱温:25℃进样体积:20μL检测器:ELSD蒸发光散射检测器http://ng1.17img.cn/bbsfiles/images/2010/11/201011291710_262707_801_3.jpg益母草供试品含量测定色谱图(主峰保留时间:22.697min)
看到一篇新闻说益母草制剂等9个批次产品不合格,我想了解一下,益母草制剂算是天然药吗?益母草制剂和益母草膏应该如何检测?处理样品的时候有没有什么要注意的?
10版药典刚加入用蒸发光检测器测益母草药材中盐酸水苏碱含量,小弟不才,死活测不出来,特此求教各位大虾的帮助。第一次测时对照色谱图比较清晰,出峰时间是2min。。。到第二次测时,对照色谱图直接成了锯齿状。此外两次测得的样品图谱都是锯齿状的,什么都看不出来。 我用的蒸发光检测器是varain-380,流动相及样品处理方法是按药典来的。泵流速0.5ml/min,柱温20℃,蒸发温度90℃,雾化温度60℃,气体(氮气)流速1.5。不知道有哪位大虾对这个比较熟悉或是做过益母草含量的,望指点迷津!有用别的检测器测益母草药材中盐酸水苏碱含量的也希望交流学习用,分光光度法就免了 再次小弟先谢谢各位大虾们了!
尊敬的专家先生: “近红外成份测定仪”这类仪器能否测定牧草(例如紫花苜蓿)中的蛋白质、脂肪等成份的含量?谢谢!
我们最近在用2010版药典方法做益母草盐酸水苏碱含量时,对照在3分多钟出了倒峰,不知道有朋友做过这个东西没有,供试品就分不开。条件:色谱条件与系统适用性试验 用丙基酰胺键合硅胶为填充剂;乙腈-0.2%冰醋酸溶液(80:20) 为流动相;用蒸发光散射检测器检测。理论板数按盐酸水苏碱计算应不低于6000。
跪求益母草 燀苦杏仁提取物鉴别图谱!!
益母草具有祛瘀生新,调经活血,素有“经产良药”之称。益母草主要有效成益母草碱、盐酸水苏碱等,益母草中盐酸水苏碱的含量测定方法较多,主要有分光光度法[1],高效液相色谱法[2,3],薄层扫描法[4]等。《中国药典》2000年版 一部[1]益母草项下对益母草规定了总生物碱的含量,采用的测定方法是分光光度法,但该方法的重现性不好。笔者曾参考文献资料报道[2,3],采用高效液相色谱法试验,文献记载中所用的条件也均未能有较好的重现性,《中国药典》 2005年版 一部[1]益母草项下的含量测定方法采用了双波长薄层色谱扫描法,但其展开剂为乙酸乙酯-正丁醇-盐酸(1:8:3),展开速度极慢,本试验对所用展开剂及显色剂、显色方法加以改进,取得满意结果,现报告如下。 1 仪器、药品与试剂 CS-930型双波长薄层扫描仪,日本岛津公司生产;硅胶G薄层预制板,青岛海洋化工厂分厂生产;盐酸水苏碱对照品(含量测定用,批号:110712-200306),购自中国药品生物制品检定所;所用试剂均为分析纯;益母草(Leonurus japonicus Houtt.)药材购自山西太原万民大药房(经山西中医学院中药鉴定教研室鉴定)。 2 方法与结果 2.1 供试品溶液的制备 取益母草0.5g,精密称定,置具塞三角瓶中,精密加入乙醇50ml,称定重量,超声处理30min,放冷,再称定重量,用乙醇补足减失的重量,摇匀,滤过,精密量取续滤液25ml,置蒸发皿中,于水浴上蒸干,用盐酸-甲醇(1:100)溶解并转移至5ml量瓶内,加盐酸-甲醇(1:100)至刻度,摇匀,作为供试品溶液。 2.2 对照品溶液的制备 精密称取经105℃干燥至恒重的盐酸水苏碱对照品20mg至20ml量瓶中,加盐酸-甲醇(1:100)溶液使溶解,并稀释至刻度,摇匀,即得(每1ml中含盐酸水苏碱1mg),作为对照品溶液。再分别取1ml,2.5ml,5 ml,7.5ml用盐酸-甲醇(1:100)溶液稀释至10ml,即得到0.1mg/ml,0.25mg/ml,0.5 mg/ml,0.75mg/ml的对照品溶液。 2.3 展开、显色及扫描条件选择 分别吸取盐酸水苏碱对照品溶液及供试品溶液,点于同一硅胶G薄层板上,以丙酮-无水乙醇-盐酸(10:10:1)为展开剂,展开,取出,晾干,于100℃加热10min,取出,放冷,喷以稀碘化铋钾-1%三氯化铁乙醇(2:1)试液,冷风吹至斑点显色清晰。进行光谱扫描,最大吸收波长λs=510mm,参比波长λR=700nm,SX=3。 2.4 线性关系的考察 精密吸取不同浓度(0.1mg/ml,0.25mg/ml,0.5mg/ml,0.75mg/ml,1mg/ml)的对照品溶液各5μl,分别点于同一硅胶G薄层板上,以丙酮-无水乙醇-盐酸(10:10:1)为展开剂,展开,取出,晾干,于100℃加热10min,取出,放冷,喷以稀碘化铋钾-1%三氯化铁乙醇(2:1)试液,冷风吹至斑点显色清晰。照薄层色谱法(《中国药典》 2000年版 一部 附录Ⅵ B)进行扫描, 以点样量为横坐标,吸收度积分值为纵坐标作图,得一直线,回归方程:Y=10869X-1973.9,r=0.9991,盐酸水苏碱点样量在1.0~10.0μg范围内呈良好线性。 2.5 精密度试验 2.5.1 异板精密度试验 精密吸取盐酸水苏碱对照品溶液4μl,分别在不同的硅胶G薄层板上点样,展开,晾干,加热,放冷,显色,扫描,测定吸收度积分值RSD%=2.15。 2.5.2 同板精密度试验 分别精密吸取盐酸水苏碱对照品溶液4μl,在同一块硅胶G薄层板上点5点,展开,取出,晾干,加热,放冷,显色,扫描,测定吸收度积分值RSD%=0.82。 2.6 稳定性试验 取供试品溶液在0、0.5、1.5、2.0h分别点样5μl ,测定样品中盐酸水苏碱斑点吸收峰面积积分值,RSD%为2.61。试验结果表明,经显色后斑点在2h内稳定。 2.7 重复性试验 按拟定的含量测定方法,对同一批样品分别制备5份供试品溶液,精密吸取5μl,点样, 测定斑点吸收峰面积积分值并计算含量。益母草中盐酸水苏碱的含量平均为0.10,RSD=1.90 %。 2.8 回收率试验 采用加样回收法试验。精密称取已知含量的益母草约0.25 g, 精密加入盐酸水苏碱对照品溶液(1mg/ml)1ml,按样品测定项下操作,计算回收率。结果盐酸水苏碱的平均回收率为97.24%, RSD%为1.75%(n=5)。 2.9 样品测定 精密吸取对照品溶液2μl、6μl和供试品溶液10μl,交叉点于同一硅胶G薄层板上,按上述条件测定斑点吸收峰面积的积分值,以外标二点法计算含量,结果益母草中盐酸水苏碱的平均含量为0.50%,RSD为2.39%(n =6)。 3 讨论 《中国药典》2000年版一部[1]益母草项下对益母草规定了总生物碱的含量,采用的测定方法是分光光度法,但无法重复。笔者曾对文献记载的高效液相色谱法所用的条件反复试验,均未能有较好的重现性。彭维[5]明确提出所有报道益母草含量测定中所用的高效液相色谱法,没有方法能够重复出来。本试验采用双波长薄层色谱扫描法,并对展开剂、显色剂及显色条件加以改进,结果满意。可为益母草及其制剂的含量测定提供有效的方法。 《中国药典》 2005年版一部[1]益母草含量测定项下展开剂为乙酸乙酯-正丁醇-盐酸(1:8:3),但展开速度缓慢,本实验以丙酮-无水乙醇-盐酸(10:10:1)为展开剂展开,速度快,分离效果好。 取供试品溶液及对照品溶液点样,展开,取出之后,必需加热至展开剂挥尽再显色,斑点在2h内稳定,否则影响显色效果。 【参考文献】 1 国家药典委员会.中华人民共和国药典.北京:化学工业出版社,2000,一部,237-238;203-204. 2 戚建中. 产妇康颗粒中盐酸水苏碱的HPLC分析.中成药, 2001,23(1):16-18. 3 姜舜尧.益母草药材中水苏碱成分的高效液相色谱法分析.药物分析杂志,2001,(4):21. 4 章曙丹. 薄层扫描法测定鲜母益胶囊中盐酸水苏碱的含量.中成药,2004,26(4):附7. 5 彭维. 产妇康颗粒质量标准研究.中药材,2003,26(3):198-200. (编辑:宋 冰) 作者单位: 030024 山西太原,山西中医学院中药系
[size=14px] [/size] [size=14px]“情志致病”是中医病因学的重要组成部分,指情志不畅会导致疾病的发生。随着现代生活节奏加快,工作压力不断增加,情志应激与疾病的关联越来越受到重视。PD发病主要与遗传内因和环境外因有关,有相同PD基因型的个体在不同的情志状态,对PD的“易感性”不同。[/size] [size=14px]从天麻钩藤饮(中医治疗PD的经典名方)化合物库中筛选出能够抑制ALOX15与PEBP1结合的天然化合物—益母草碱(Leo),并发现Leo能明显缓解情志应激加剧的PD样行为障碍,降低PD易感性。[/size] [size=14px] [/size] [size=14px]该研究通过拘束应激模拟“情志失调”诱导肝损伤的发生,发现拘束应激制造成生物节律的失调,其中主要引起生物节律因子BMAL1发生显著动荡,增加其表达水平。随后研究发现,BMAL1可直接与磷脂过氧化调节的重要酶ALOX15的启动子序列结合,促进其表达,致使以氧化磷脂(oxPLs)为代表的大量氧化脂质产物堆积。而肝细胞的大量磷脂过氧化状态,是一种高势能、高熵值的不稳定状态,会导致对病毒、肿瘤等重大疾病易感性增加。此外,片仔癀属于清热类名贵中成药,药理作用广泛,临床上多应用于保肝护肝,该研究在解释情志应激诱导肝脏损伤的作用机制的基础上,表征了片仔癀调节BMAL1-ALOX15通路,降低情志应激小鼠肝脏中的脂质过氧化水平,缓解肝损伤。[/size] [size=14px] [/size] [size=14px]1、应激诱导的磷脂过氧化作用增强PD易感性[/size] [size=14px]为了探究生理应激对PD的影响,作者首先从PD模型(hSNCAA53T转基因小鼠)出发,通过拘束应激模拟“情志失调”,发现慢性拘束应激模拟的情志应激状态能显著缩短A53T小鼠的发病时间。此外,在压力后A53T小鼠的中脑脂质过氧化终产物4-HNE和MDA的水平均升高,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS的磷脂组学也显示应激A53T小鼠的中脑存在广泛的磷脂过氧化,其中铁死亡标记物磷脂酰乙醇胺过氧化物(ox-PEs)最为突出,压力还改变了铁死亡途径中涉及的蛋白质和基因,这些结果表明压力可能通过磷脂过氧化增加PD敏感性(图1)。[/size] [size=14px]图片[/size] [size=14px]图1 应激诱导的磷脂过氧化作用增强PD易感性[/size] [size=14px]2、应激引起的CORT升高有助于磷脂过氧化和PD易感性[/size] [size=14px]与啮齿动物的中枢应激反应系统(由HPA轴控制)通常通过释放皮质类固醇做出适应性反应的理论一致,激素组学分析的结果表明,小鼠受到压力后CORT(皮质酮)是最显著改变的激素。且在A53T小鼠中CORT加剧了行为障碍、多巴胺能神经元损失等表型,且增加4-HNE和MDA以及磷脂过氧化,改变了参与铁死亡相关途径的蛋白质和基因,与压力引起的类似。这些结果表明CORT可能在应激过程中促进磷脂过氧化发挥重要作用,进而导致PD的易感性(图2)。[/size] [size=14px]图片[/size] [size=14px]图2 应激引起的CORT升高有助于磷脂过氧化和PD易感性[/size] [size=14px]3、CORT诱导的磷脂过氧化是由ALOX15与PEBP1结合介导的[/size] [size=14px]PE结合蛋白1(PEBP1)通常与Raf-1激酶结合并抑制Raf-1激活的MEK/ERK途径,而PKC磷酸化会导致PEBP1从Raf-1解离以及下游蛋白的激活。有证据表明,Raf-1释放的PEBP1与15-脂氧合酶-1(ALOX15)结合并形成复合物,催化含有多不饱和脂肪酸的膜脂的氧化,进而引发铁死亡细胞。作者发现A53T小鼠表现出ALOX15表达的显著上调,这与压力无关。在应激的WT和A53T小鼠中观察到PEBP1磷酸化显著增加,表明PEBP1可能与Raf-1解离。考虑到在接受应激或CORT处理的小鼠中脑中观察到磷脂过氧化物的显著积累,作者研究了ALOX15/PEBP1复合物的变化,发现CORT 显著增强了PEBP1和ALOX15之间的相互作用,而Raf-1/PEBP1复合物解离,且应激诱导显示出和CORT 类似效果。这些发现表明A53T诱导的ALOX15上调和CORT诱导的PEBP1解离可能是随后脂质过氧化的原因(图3)。[/size] [size=14px]图片[/size] [size=14px]图3 CORT诱导的磷脂过氧化是由ALOX15与PEBP1结合介导的[/size] [size=14px]4、ALOX15对于应激诱发的PD易感性至关重要[/size] [size=14px]有研究报道铁死亡抑制剂ferrostatin-1(Fer-1)能够通过自由基捕获和干扰ALOX15/PEBP1形成来抑制磷脂过氧化。作者发现Fer-1逆转了A53T小鼠应激诱导的PD易感性,减弱了应激诱导的脂质过氧化和铁死亡信号通路激活,降低了应激诱导的ALOX15/PEBP1复合物的水平,表明多巴胺能神经元的铁死亡变性可能在PD易感性增强中发挥着至关重要的作用。由于ALOX15对于膜中的脂质过氧化至关重要,作者采用Alox15敲除小鼠发现ALOX15敲除显著减弱了与 A53T 和压力相关的PD运动和协调功能受损,且对多巴胺能神经元的损伤显著减少,中脑中因过度表达的A53T和应激而出现的脂质过氧化水平较低,表明ALOX15/PEBP1在PD小鼠模型中应激增强的易感性中发挥着至关重要的作用(图4)。[/size] [size=14px]图片[/size] [size=14px]图4 ALOX15对于应激诱发的PD易感性至关重要[/size] [size=14px]5、益母草碱抑制ALOX15与PEBP1结合从而抑制铁死亡[/size] [size=14px]作者之前发现天麻钩藤颗粒(TG)的中药制剂通过抑制ALOX15介导的脂质过氧化来减轻PD模型的行为障碍,为了发现潜在的活性化合物,作者将TG的主要成分与ALOX15对接,发现在所研究的17种化合物中益母草碱通过与靠近PEBP1结合位点的Arg166、Glu169、Lys172和Phe167 形成氢键,表现出与ALOX15 互作的最大潜力。此外,通过MST、CETSA也证明益母草碱直接结合ALOX15而不是PEBP1。但是,益母草碱不会抑制ALOX15酶活性,而是破坏ALOX15/PEBP1相互作用,因此,即使在ALOX15过表达时,益母草碱剂量依赖性地挽救了RSL3诱导的铁死亡和脂质过氧化。此外,益母草碱显著逆转CORT诱导脂质过氧化产物4-HNE和MDA增加,以及TFRC和Ptgs2激活,这些结果表明益母草碱抑制ALOX15与PEBP1结合从而抑制铁死亡(图5)。[/size] [size=14px]图片[/size] [size=14px]图5 益母草碱抑制ALOX15与PEBP1结合从而抑制铁死亡[/size] [size=14px]6、益母草碱减轻应激小鼠对PD的易感性[/size] [size=14px]最后,作者采用MPTP诱导的PD小鼠模型和先前描述的PD易感小鼠模型评价益母草碱的疗效,发现益母草碱治疗能显著改善小鼠运动协调性,逆转多巴胺能神经元损失和与PD相关的脂质过氧化。进一步研究了益母草碱对小鼠体内ALOX15和PEBP1结合的影响,发现益母草碱显著降低了ALOX15和PEBP1的结合,同时倾向于增加PEBP1与Raf-1的结合。此外,益母草碱显著减少A53T和应激引起的膜脂过氧化物的积累,降低TFRC和Ptgs2的水平。研究结果表明益母草碱可能通过抑制脂质过氧化来降低小鼠对PD的易感性(图6)。[/size] [size=14px]图片[/size] [size=14px]图6 益母草碱减轻应激小鼠对PD的易感性[/size] [size=14px]总结[/size] [size=14px]拘束应激激活HPA轴释放皮质激素,其中,皮质酮(CORT)变化最为显著。随后,CORT激活蛋白激酶PKC通路,促使PE结合蛋白PEBP1发生磷酸化从而从Raf-1上解离,游离的PEBP1与脂氧合酶ALOX15形成强有力的过氧化磷脂的催化器,致使以oxPEs为代表的大量的氧化脂质产物的堆积,增加DA神经元对铁死亡(ferroptosis)的易感性,并最终造成PD的发生和发展。因此,ALXO15-PEBP1可能作为PD治疗药物开发的潜在靶点,中药复方天麻钩藤饮中的活性成分益母草碱能有效抑制ALXO15-PEBP1结合,延缓神经元的脂质氧化和退行性丢失(图 7)。[/size]
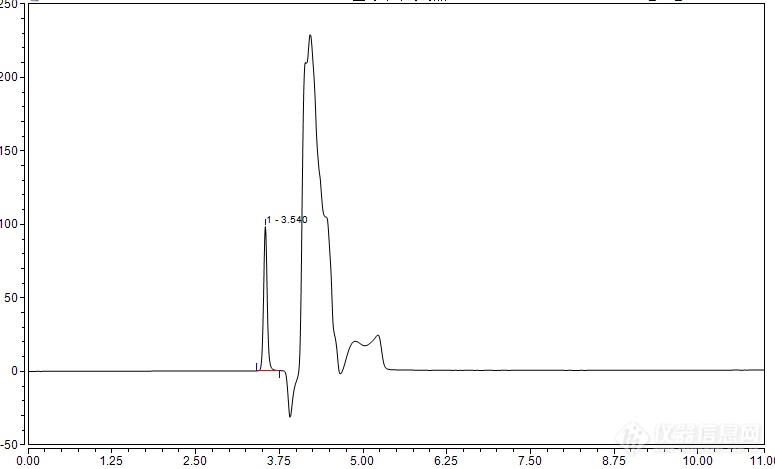
问题好多,烦请各位老师认真看一下益母草颗粒或益母草膏中盐酸水苏碱含量测定,色谱条件:乙腈-0.05mol/l磷酸二氢钾-磷酸(15:85:0.15)为流动相。色谱柱为Biobasic SCX,Thremofisher公司的。液相色谱仪为赛默飞世尔U3000高效液相色谱仪。柱温30,流速1.0ml/min。检测波长为192nm,在此条件下检测不出色谱峰,后将0.05mol/l的磷酸二氢钾改为0.03mol/l的浓度,结果出峰了,但峰的分离效果不好图1是对照的色谱峰,对照是用甲醇配制的,然后我进了一针甲醇的空白结果如图2可以看出加了对照后,第一个色谱峰变大,后面多了一个峰,不过没分开。为了确认该峰是否为要的峰,我在对照品溶液中又加了一点对照品,结果如图3前后两个峰都变大,郁闷死我了。。。。。。。。。。。期间也调低了流速,调低了柱温,更改了流动相中乙腈的比例,结果都不理想,后来将0.05mol/l的磷酸二氢钾改为0.01mol/l的浓度,对照品的峰可以比较明显的分出来了,如图4但是峰形不好,本想这样将就用着的,后来在这个对照中加入一点对照品后,前后峰的面积又都变大了。峰如下图5这是什么原因呢?是盐酸水苏碱溶解在甲醇中产生了什么反应么?为什么甲醇中也有那个色谱峰呢?之后我决定进样品,益母草颗粒,称一克,用0.5%的盐酸甲醇溶液25ml超声30min,进样后结果如图6问题好多啊,为什么对照峰前拖尾,样品峰后拖尾?还有样品我也尝试了那些方法,都没有使峰很好的分离出来,样品该怎么分离好呢?还得调磷酸二氢钾浓度么?之前看到网上有很多说用乙腈-0.05mol/l磷酸二氢钾-磷酸(15:85:0.15)的流动相做出结果的,为什么我的做不出来,要换成0.01mol/l的呢?[img=,690,417]https://ng1.17img.cn/bbsfiles/images/2019/08/201908131150569283_4003_1791505_3.png!w690x417.jpg[/img][img=,690,422]https://ng1.17img.cn/bbsfiles/images/2019/08/201908131150586305_6177_1791505_3.png!w690x422.jpg[/img][img=,690,426]https://ng1.17img.cn/bbsfiles/images/2019/08/201908131151014335_4399_1791505_3.png!w690x426.jpg[/img][img=,690,428]https://ng1.17img.cn/bbsfiles/images/2019/08/201908131151040285_6173_1791505_3.png!w690x428.jpg[/img][img=,690,420]https://ng1.17img.cn/bbsfiles/images/2019/08/201908131151065795_1809_1791505_3.png!w690x420.jpg[/img][img=,690,413]https://ng1.17img.cn/bbsfiles/images/2019/08/201908131151088295_2442_1791505_3.png!w690x413.jpg[/img]
益母草与川芎是中药成方制剂中配伍应用频率较高的药对,益母草主要含水苏碱等生物碱类活性成分,川芎主要含阿魏酸等活性成分。对益母草药材及其制剂《中国药典》常以雷氏盐剩余比色法测定益母草总碱含量。在对含益母草及川芎这一药对的制剂进行质量控制时,由于川芎所含生物碱也能与雷氏盐反应生成沉淀,且雷氏盐剩余比色法本身重现性较差,故不宜采用该法。为此,本实验建立了以薄层扫描法测定益母草与川芎合煎液中盐酸水苏碱含量的方法,为制订含有益母草与川芎药对制剂的质量标准提供参考。 1 仪器与试药 CS-9000型双波长薄层扫描仪(日本岛津):939薄层制板器(重庆南岸贝尔德仪器技术厂);定量毛细管(Drummond USA)。盐酸水苏碱对照品(中国药品生物制品检定所,712200105);益母草、川芎(购于安徽毫州)。硅胶G(化学纯,青岛市北区海化干燥剂厂);强酸性阳离子交换树脂(732型,上海化学试剂采购供应站);其余试剂均为分析纯。 2 方法与结果 2.1 益母草与川芎合煎液的制备 取药材100g(益母草-川芎:1:1),加水煎煮2次,每次2h,每次加12倍量水,合并煎液,滤过,滤液浓缩并定容至100mL,即得合煎液样品。 2.2 对照品溶液的制备 取105℃干燥至恒重的盐酸水苏碱对照品适量,精密称定,加乙醇溶解定容,制成1.2mg/mL的对照品溶液。 2.3 供试品溶液的制备 取合煎液样品适量,离心(4O00r/min)5min后,精密量取上清液lmL置小烧杯中,加蒸馏水1OmL,用稀盐酸调pH 1~2,通过已处理好的强酸性阳离子交换树脂柱(内径1cm,长1Ocm)用水洗至流出液无色,弃去水液,再以乙醇-氨水(8:2)150mL洗脱,收集洗脱液,蒸干,残渣加乙醇溶解,定量转移并定容于lmL容量瓶中,摇匀,作为供试品溶液。 2.4 薄层色谱与扫描条件 吸附剂:硅胶G-O.5%CMC-Na薄层板(厚约0.5mm,105℃活化lh,置干燥器中备用):展开剂:丙酮可-无水乙醇-盐酸(10:6:1) :显色剂:喷以改良碘化铋钾-1%FeC13无水乙醇液(5:1)。,冷风吹至斑点显色清晰。 描方式:双波长反射式锯齿扫描:检测波长:λs=525nm,λR=660nm:狭缝:0.4mm×0.4mm: 线形参数SX=3。 2.5 标准曲线的制备 分别精密吸取盐酸水苏碱对照品溶液4.0、6.0、8.0、10.0、12.0μL,分别点于同一薄层板 ,依上述条件展丌,显色,在薄层板上覆盖同样大小的玻璃板,周围用胶布吲定,然后扫描测定各斑点的峰面积积分值。以峰面积积分值(A)对点样量(C)进行回归,得回归方程:A=-37152.7+7579.18C‘r=O.9986。表明在4.8~14.4μg范围内盐酸水苏碱斑点峰面积积分值与点样量呈良好的线性关系。 2.6 稳定性考察 取供试品溶液5μL点样,依上述条件展开,显色,在1h内每隔10min扫描测定1次,结果斑点峰面积积分值RSD为4.6%(n=6),表明斑点在显色后1h内基本稳定。 2.7 精密度考察 精密吸取供试品溶液在同一薄层板上点相同量5点,每点5μL,按上述条件展开,显色,扫描测定,斑点峰面积秋分值RSD为1.33%(n=5),对其中一点连续扫描测定5次,斑点峰面积积分值RSD为0.62%(n=5),表明同板精密度和仪器精密度较好。 2.8 异板精密度试验 取薄层板3块,在每1块薄层板二分别点对照品溶液4、6μL及供试品溶液5μL,依法展开,显色,扫描测定,采用外标两点法计算含量。结果测得供试品溶液中水苏碱含量为1.1308 mg/mL(RSD=3.01%)。 2.9 回收率测定 精密量取已知含量的合煎液样品0.5mL,加入盐酸水苏碱对照品溶液0.5mL,混匀后,按2.3项方法制备供试品溶液。精密吸取供试液5μL,对照品溶液4、6μL,变又点样于同一薄层板上,按上述条件展开、显色、扫描测定,用外标两点法计算。 2.10 样品测定 取合煎液样品,按2.3项 方法制备供试品溶液。精密吸取供试品溶液5μL,对照品溶液4、6μL,分别交叉点于同一薄层板上,按上述条件展开,显色,扫描测定,用外标两点法计算样品中盐酸水苏碱的含量。 3 讨论 在供试品溶液制备中,以甲醇-氨水(8:2) 或乙醇-氨水(8:2) 作洗脱剂,提纯效果均较好。因乙醇比甲醇价廉且安全,建议采用乙醇-氨水(8:2)作为洗脱剂。本实验对洗脱剂用量考察结果表明,用14OmL即可将盐酸水苏碱洗脱完全。为了保证洗脱充分,本实验中确定用150mL洗脱。 盐酸水苏碱为水溶件物质,在薄层板上显色时受湿度影响很大。通过实验考察,优选出其较好的显色方式为:展开后,取出,晾干,丁105℃烘lOmin,再喷显色剂,冷风吹至斑点色清晰。 益母草有效成分水苏碱含量较低且波动大,容易造成含益母草的制剂有效成分低、产品质量不稳定 ,应增加水苏碱含量控制项目。本实验建立的含量测定方法可为制订含益母草及川芎制剂中水苏碱的定量标准提供参考。参考文献:[1]国家药典委员会.中国药典(一部)[S].北京:化学工业出版社,2000.237,455,564.[2]张玲,时延增,于宗渊,等.舣波长薄层扫描法测定益母草LJ服液中水苏碱的含量[J].中国药科大学学报,1996,27(1):16—18.[3]张玲, 宗渊,李国宝,等.7种含益母草中成药中水苏碱的含量测定[J].药物分析杂志,1996,16(3):181—183.

作者:曾中强(怀化市中医院药剂科,湖南怀化,418000)摘要: 目的:建立高效液相色谱法测定益母草药材及其颗粒制剂中水苏碱含量的方法.方法:色谱柱为Diamonsil C18(4.6mm×250mm);流动相为甲醇-0.05mol/L磷酸二氢钾溶液(70:30);检测波长210 nm,柱温25℃,流速1.0mL/min.结果:益母草药材及其制剂中水苏碱的保留时间均为15.1 min,色谱峰分离良好,对照品水苏碱线性范围为1.01~30.39 ug/mL(R2=O.9997,n=5),方法回收率为100.1%,RSD为1.03%。结论:该法可用于益母草流浸膏中及其颗粒制剂中水苏碱的含量测定,可以用来对提取物及相关制剂进行质量控制。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208071358_382237_1609970_3.jpg
今天去抽检,厂家说益母草颗粒剂的有效成分随着时间变长,含量也降低。是什么原因呢,难道没有办法解决这个问题吗?
[size=20px][color=#93c6bc][b]鉴别[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [font=宋体][/font][b][font=宋体][/font][/b][font=宋体][/font] [b][font=宋体][/font][/b][font=宋体][/font][font=宋体][/font] [font=宋体](1)本品茎横切面:表皮细胞外被角质层,有茸毛;腺鳞头部4、6细胞或8细胞,柄单细胞;非腺毛1[/font][font=&]~[/font][font=宋体]4[/font][font=宋体]细胞。下皮厚角细胞在棱角处较多。皮层为数列薄壁细胞;[color=var(--weui-LINK)]内皮层[i][/i][/color]明显。中柱鞘纤维束微木化。韧皮部较窄。木质部在棱角处较发达。髓部薄壁细胞较大。薄壁细胞含细小草酸钙针晶和小方晶。鲜品近表皮部分皮层薄壁细胞含叶绿体。[/font] [font=宋体](2)取[color=var(--weui-LINK)]盐酸水苏碱[i][/i][/color]〔含量测定〕项下的供试品溶液10ml,蒸干,残渣加无水乙醇1ml使溶解,离心,取上清液作为供试品溶液(鲜品干燥后粉碎,同法制成)。另取盐酸水苏碱对照品,加无水乙醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各5[/font][font=&]~[/font][font=宋体]10[/font][font=宋体]μl,分别点于同一硅胶G薄层板上,以丙酮-无水乙醇-盐酸(10:6:1)为展开剂,展开,取出,晾干,在105℃加热15分钟,放冷,喷以稀碘化铋钾试液-三氯化铁试液(10:1)混合溶液至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。[/font] [font=宋体][/font][font=宋体][/font] [font=宋体][/font] [size=20px][color=#93c6bc][b]检查[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [b][font=宋体][/font][font=宋体] [/font] [font=宋体][/font] [font=宋体]水分[/font][/b][font=宋体] [/font][font=宋体]干益母草 不得过13.0%(通则0832第二法)。[/font] [b][font=宋体]总灰分[/font][/b][font=宋体] [/font][font=宋体]干益母草 不得过11.0%(通则2302)。[/font] [b][font=宋体]【浸出物】 [/font][/b][font=宋体]干益母草 照水溶性浸出物测定法(通则2201)项下的热浸法测定,不得少于15.0%。[/font] [b][font=宋体]【含量测定】[/font][font=宋体] 干益母草 盐酸水苏碱 [/font][/b][font=宋体]照高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法(通则0512)测定。[/font] [b][font=宋体]色谱条件与系统适用性试验 [/font][/b][font=宋体]以丙基酰胺键合硅胶为填充剂;以乙腈-0.2%[color=var(--weui-LINK)]冰醋酸溶液[i][/i][/color](80:20)为流动相;用蒸发光散射检测器检测。理论板数按盐酸水苏碱峰计算应不低于6000。[/font] [b][font=宋体]对照品溶液的制备 [/font][/b][font=宋体]取盐酸水苏碱对照品适量,精密称定,加70%乙醇制成每1ml含0.5mg的溶液,即得。[/font] [b][font=宋体]供试品溶液的制备 [/font][/b][font=宋体]取本品粉末(过三号筛)约1g,精密称定,置具塞锥形瓶中,精密加入70%乙醇25ml,称定重量,加热回流2小时,放冷,再称定重量,用70%乙醇补足减失的重量,摇匀,滤过,取续滤液,即得。[/font] [b][font=宋体]测定法 [/font][/b][font=宋体]分别精密吸取对照品溶液5μl、10μl,供试品溶液10[/font][font=&]~[/font][font=宋体]20[/font][font=宋体]μl,注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],测定,用外标两点法对数方程计算,即得。[/font] [font=宋体]本品按干燥品计算,含盐酸水苏碱(C[sub]7[/sub]H[sub]13[/sub]NO[sub]2[/sub]?HC1)不得少于0.50%。[/font] [b][font=宋体]盐酸益母草碱[/font][/b][font=宋体] [/font][font=宋体]照高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法(通则0512)测定。[/font] [b][font=宋体]色谱条件与系统适用性试验 [/font][/b][font=宋体]以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.4%辛烷磺酸钠的0.1%磷酸溶液(24:76)为流动相;检测波长为277nm。理论板数按盐酸益母草碱峰计算应不低于6000。[/font] [b][font=宋体]对照品溶液的制备 [/font][/b][font=宋体]取盐酸益母草碱对照品适量,精密称定,加70%乙醇制成每1ml含30μg的溶液,即得。[/font] [b][font=宋体]测定法 [/font][/b][font=宋体]分别精密吸取对照品溶液与盐酸水苏碱〔含量测定〕项下供试品溶液各10μl,注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],测定,即得。[/font] [font=宋体]本品按干燥品计算,含盐酸益母草碱(C[sub]14[/sub]H[sub]21[/sub]O[sub]5[/sub]N[sub]3[/sub]?HC1)不得少于0.050%。[/font] [font=宋体] [/font]
[b]《中国药典》2010年版一部益母草颗粒增加了盐酸水苏碱的含量测定,是采用薄层扫描法。本人做含测时,碰到了一个奇怪的事情:有的时候显色后只见到8微升对照液的斑点,而3微升对照液和样品斑点不出来;有的时候又能出来,不知咋回事?展开剂是:丙酮-无水乙醇-盐酸(10:6:1)。请教各路英雄,问题出在哪里?[/b]
请问益母草做盐酸水苏碱时的蒸发光检测器的温度和气流量是多少?
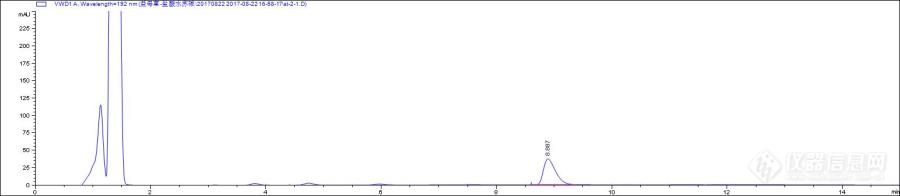
[b]小序:益母草中盐酸水苏碱含量测定采用的是蒸发光检测器,蒸发光检测器受气体纯度及流动相影响较大,样品复杂,检测器容易被污染,实验室建立了另外一种上机条件与大家分享。[/b]1 材 料 益母草药材(送检样);乙醇(分析纯),乙腈(色谱级),乙酸;[color=#333333]盐酸水苏碱[/color][color=#333333]对照品[/color][color=#333333](购自中检院[/color][color=#333333])[/color]。[color=#333333]2 仪器与设备[/color][color=#333333] 岛津液相LC-20AT[/color][color=#333333]配制奥泰ELSD蒸发光检测器,安捷伦1260 InfinityII 色谱仪,[/color][color=#333333]色谱柱安捷伦[/color][color=#333333]Hilic plus (10[/color][color=#333333]0mm*4.6μm*[/color][color=#333333]3.5[/color][color=#333333]μm);[/color]DZKW-S-6型电热恒温水浴锅(北京市永光明医疗器械仪器有限公司)。[color=#333333]3 实验方法[/color][color=#333333]3.1 色谱条件[/color][color=#333333] [/color][color=#333333] 条件1:紫外检测器,[/color][color=#333333]波长192nm(末端吸收),[/color][color=#333333]流动相:乙腈:[/color][color=#333333]水(80:20),[/color][color=#333333]柱温35℃,[/color][color=#333333]进样量:5[/color][color=#333333]μL;[/color][color=#333333] 条件2:蒸发光检测器,漂移管温度105[/color][color=#333333]℃,载气(氮气)流速:2.5L/min,流动相:[/color][color=#333333]乙腈:0.2%冰醋酸溶液[/color][color=#333333](80:20)。[/color][color=#333333]3.1 对照品溶液的制备[/color][color=#333333] 取盐酸水苏碱对照品适量,精密称定,加70%乙醇制成每1ml含[/color][color=#333333]0.6 [/color][color=#333333]mg的溶液,即得[/color][align=center][color=#333333][img=,569,165]https://ng1.17img.cn/bbsfiles/images/2019/07/201907231136427972_5007_1858223_3.png!w569x165.jpg[/img][/color][/align][align=center][color=#333333]图1 盐酸水苏碱标品紫外检测器色谱图[/color][/align][align=center][color=#333333][img=,586,192]https://ng1.17img.cn/bbsfiles/images/2019/07/201907231137411988_3077_1858223_3.png!w586x192.jpg[/img][/color][/align][align=center][color=#333333][color=#333333]图2 盐酸水苏碱标品蒸发光检测器色谱图[/color][/color][/align][color=#333333]3.2 供试品溶液的制备 [/color][color=#333333] 取本品粉末(过三号筛)约1[/color][color=#333333] [/color][color=#333333]g,精密称定,置具塞锥形瓶中,精密加入70%乙醇25[/color][color=#333333] [/color][color=#333333]m[/color][color=#333333]L[/color][color=#333333],称定重量,加热回流2小时,放冷,再称定重量,用70%乙醇补足减失的重量,摇匀,滤过,取续滤液,即得。[/color][align=center][color=#333333][img=,578,172]https://ng1.17img.cn/bbsfiles/images/2019/07/201907231138187006_8379_1858223_3.png!w578x172.jpg[/img][/color][/align][align=center][color=#333333]图3 益母草样品紫外检测器色谱图[/color][/align][align=center][color=#333333][img=,549,161]https://ng1.17img.cn/bbsfiles/images/2019/07/201907231139006558_7913_1858223_3.png!w549x161.jpg[/img][/color][/align][align=center][color=#333333][color=#333333]图4 益母草样品蒸发光检测器色谱图[/color][/color][/align]4 结果与讨论 两种检测器测得的结果一致性良好,蒸发光检测器需要氮气做为载气,为了节约成本我们开发用紫外检测器进行检测,波长虽然是末端吸收容易受杂质干扰,但是选择合适的流动相比例有利于去除这些杂质干扰,使样品中的盐酸水苏碱得到很好的分离,并且可以准确定量。
据新西兰初级产业部(MPI)消息,针对新西兰牛奶中检出低含量双氰胺的报道,新西兰两家最大的肥料公司已暂停双氰胺的销售,新西兰初级产业部对两家公司采取的行动表示支持。 双氰胺是肥料辅助品,用于牧场可增加牧草的产量,还可防止肥料的副产品硝酸盐流入河流和湖泊,并减少温室气体的排放。 新西兰初级产业部表示,目前双氰胺在食品中的限量尚无国际标准,使用双氰胺不存在食品安全风险,而且双氰胺毒性较低,未发现高剂量使用会出现不良反应。 然而新西兰MPI担心,由于目前尚无国际标准,消费者不能接受牛奶中检出双氰胺的事实,这可能会对新西兰的乳品贸易产生影响。 原文链接:据了解,现在有很多第三方机构及政府机构提供双氰胺检测的方法有HPLC \LC-MS\LC-MS-MS ,后者居多,本机构也是提供LC-MS-MS 检测方法检测
苜蓿(mùxu)是苜蓿属(Medicago)植物的通称,俗称金花菜,是一种多年生开花植物。其中最著名的是作为牧草的紫花苜蓿(Medicago sativa)。苜蓿种类繁多,多是野生的草本植物。中国产的苜蓿主要有三种。营养价值很高,具有清脾胃、利大小肠、下膀胱结石的功效。 苜蓿含有最丰富的维他命K,成分之高,驾乎一切蔬菜之上。其他如维他命C、B也相当丰富。多为牲畜饲料。在[url=https://baike.so.com/doc/5417629-5655777.html]西汉[/url]从[url=https://baike.so.com/doc/5415894-5654039.html]西域[/url]引进。
[b]要有效遏制鼠害,必须解决超载过牧,这就要运用生态智慧,调整人与自然的关系,杜绝对自然的过度索取[/b] 提及草原,人们常会想到那句著名的古诗:“天苍苍,野茫茫,风吹草低见牛羊。”如今,这样的风景已很难再觅。 5月的内蒙古草原,正遭受着一场严重的鼠害:草原上满地的鼠洞,让骏马难再奔驰;刚刚返青的牧草,被老鼠大肆吞噬;绿色的草地正在变成黄褐色的荒漠。据统计,内蒙古草原鼠害危害面积超过9800万亩。 当地人说鼠害猖獗是因为草原退化,可草原退化又是因为什么呢?正如生态学所揭示的,除了气候等自然因素,主因还是人类无节制的活动——既有历史上的开垦农田,也有近年来的滥捕草原动物,还有长期的超载过牧。 草原也是人类的家园,怎么能不让人活动?关键要节制,要敬畏自然,尊重草原生态系统的规律。 内蒙古草原上,千百年来流传着一段古老的对话。孩子问母亲:“妈妈,我们为什么要不停地搬迁?”母亲说:“孩子,我们要是固定在一处,大地母亲就会疼痛,我们不停地搬迁,就像血液在流动,大地母亲就会感到舒服。”这一对话让我们看到游牧和草原生态之间那种相互依存的辩证关系。 草原生态既是美好的,也是脆弱的。在草原上,牲畜对草场的作用表现在采食、踩踏和施肥三个方面。适度的采食、踩踏、施肥可以松土并促进牧草的生长,但草原经不起牲畜长时间的频繁踩踏。所谓不能竭泽而渔,我们可以利用草原,但也要给草原以休养生息的机会。 内蒙古的浑善达克沙地是京津沙尘暴的策源地之一,那里在历史上曾经水草丰美,有“塞外江南”之美誉。上世纪80年代以后,牧民的牲畜成倍增长,过度放牧导致沙化程度愈演愈烈。9年前,中科院专家试验性地在已严重沙化的4万亩草原上禁耕、禁牧,让其自然恢复,牧民养畜所需饲草在小范围的人工高效地上生长。如今,这片草原重现生机,野兔、狐狸、大雁、灰鹤、狼等野生动物也渐次回归。 我们并不敢说,4万亩草原上的做法能够复制到所有草原,可它至少说明了一个问题,解决草原退化的问题,需要一种系统的眼光,而不能头痛医头,脚痛医脚。 当前内蒙古的鼠患,令人揪心,为解燃眉之急,投入更多的资金,施用技术含量更高的药物,非常必要;把鼠情预测预报与防治体系建设纳入国家规划,对于遏制年复一年的鼠害也是意义重大。然而,仅仅这些,似乎还难以彻底消除造成鼠害的主因——超载过牧。 解决超载过牧,根治草原鼠患,既要有应急之举,更应该运用生态智慧,调整人与自然的关系,杜绝对自然的过度索取。应从生态系统的整体恢复着眼,完善现行草原承包制度,选择合适的技术路线,科学管理和利用草原。草原之外的人也要深刻认识到,自己的衣食住行在哪些环节过度消耗了资源,造成了污染,从而克制过度的欲望,从身边做起,造福生态。
[font=微软雅黑]点击链接查看更多:[url]https://www.woyaoce.cn/service/info-4155.html[/url]乙草胺(acetochlor),即2-乙基-6甲基--N-乙氧基甲基-α-氯代乙酰替苯胺,是一种广泛应用的除草剂,人体暴露在乙草胺每日摄取容许量以上的环境下会对造成一定的潜在危害影响,并且目前还不能排除基因毒性的存在。[/font][font=微软雅黑]乙草胺因其毒性,被美国环境保护局定为B-2类致癌物,规定在1个月的监测期,在20个监测井所谓地下水中残留浓度不得超过0.1μg/L,kolpin等报道了美国在乙草胺的施用期的样品检测结果,1994年雨水和喝水中最大检出浓度分别是2.5、1.2μg/L。乙草胺作为玉米、大豆、棉花和果树的除草剂,在我国的年使用量已超多10000t(原药)。[/font][font=微软雅黑]丁草胺(Butachlor),2-氯-N-(2,6-二乙基苯基)-N-(丁氧甲基)乙酰胺,是选择性芽前除草剂,植物吸收丁草胺后,在体内抑制和破坏蛋白酶,影响蛋白质的形成,抑制杂草幼芽和幼根正常生长发育,从而使杂草死亡。在粘壤土及有机质含量较高的土壤上使用,药剂可被土壤胶体吸收,不易被淋溶,特效期可达1-2个月。[/font][font=微软雅黑]丁草胺具有挥发性,它对人体皮肤和眼睛有轻微的刺激作用,中毒的主要表现为消化系统与神经系统症状。轻者可引起胃肠功能紊乱,出现恶心、呕吐、吞咽困难、头晕、头痛等;严重者可引起麻醉作用,表现为头痛、头晕、无力、面潮红,酒醉状态,恶心、呕吐、呼吸困难、眼和呼吸道刺激症状和四肢麻木等。严重时可出现意识蒙眬、抽搐、昏迷、心室纤颤,呼吸停止而即刻死亡。[/font][font=微软雅黑]鉴科检测参考《DB21T 1546-2007 土壤中乙草胺和丁草胺残留量的测定 [url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法》,建立了利用全自动固相萃取仪(Fotector Plus)结合[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]检测沉积物中乙草胺和丁草胺残留量的方法。在40mL丙酮-正己烷(4+1)提取后过滤,再用30mL丙酮-正己烷(4+1)复提2次,合并提取液。使用Auto EVA-08IR浓缩至2mL后 Fotector Plus全自动固相萃取仪净化,自动完成 SPE 柱活化、样品上样、淋洗、收集等步骤,收集液再氮吹浓缩、溶剂转换、定容后,用GC检测。[/font]
氟噻草胺(flufenacet)是由拜耳于1998年首次推出的,氟噻草胺与苯噻酰草胺一样同属芳氧乙酰胺类化合物,与氯代乙酰胺类除草剂具有类似的杂草防除谱,可用于玉米、大豆、番茄、马铃薯、水稻等作物,防除一年生禾本科杂草、莎草和一些小粒阔叶杂草。目前,氟噻草胺主要用作土壤处理剂,芽前、芽后皆可使用。1998年氟噻草胺在美国取得全球首次登记,近年来拜耳作物科学将氟噻草胺与防治阔叶杂草的除草剂,如嗪草酮、吡氟酰草胺、异恶唑草酮、磺草唑胺及二甲戊灵等进行复配,并上市了众多产品。近几年的销售额呈逐年下降趋势,2012年的全球销售额为1.79亿美元,2013年为1.55亿美元,到2014年销售额为1.50亿美元。目前,氟噻草胺的全球原药产量在1000吨左右,全球市值超过1亿美元。美国Albaugh公司在2015年取得欧洲的登记。由于该产品被列入欧盟ANNEX I,数据保护要到2018年才到期,这也意味着在2019年之前,在欧洲市场只有拜耳和Albaugh两家公司有权销售氟噻草胺,其他公司要等数据保护结束后重新登记才可以。2015年,拜耳取得了几个临时登记,氟噻草胺在中国首次正式亮相,国内暂无氟噻草胺的正式登记。虽然氟噻草胺的化合物专利已于2009年到期,但其工艺专利仍处于有效期内。此外,氟噻草胺的生产工艺中包含多个独立步骤,其中一些工艺需要特殊的操作,涉及到技术、安全、环保等问题,这也是国内企业在实现大规模生产之前必须解决的问题。
《农用地土壤环境质量标准》是对现行《土壤环境质量标准》直接修改,适用于农田、果园、茶园、牧草地等农用地土壤环境质量评价与管理。修订草案删除了现行标准中“一刀切”规定的自然背景值(一级标准值)和高背景值(三级标准值),按照土壤pH条件将原标准规定的镉(Cd)限值由0.3mg/kg和0.6mg/kg两档细化为0.3mg/kg、0.4mg/kg、0.5mg/kg和0.6mg/kg四档,收严了铅、六六六、滴滴涕3项污染物限值,增加了总锰、总钴等10项污染物选测项目,更新了监测规范。
38.3mg/l。它的除草活性 Pentoxazone从每公顷0.15-0.45公斤有效成分的浓度于苗前和苗后早期施用,对小果子一年生杂草如稗属Oryzice-la,雨久花属Vaginalis、莎草属difformis和阔叶杂草有良好的除草效果。许多多年生蓑衣杂草如荸荠属Kuroguwai;也有控制和打击作用。在苗前和苗后早期(-4+5)应用时,杂草还未长到10叶阶段施用最有效,且充分发挥本除草剂功效;当以浓度每公顷0.39-0.45公斤有效成分使用时,Pentoxazone能迅速杀灭稗属Oryzicola并残留部分一直能控制6周,它的长久持效性是由于土壤对它吸收而具有低迁移性和水中低溶解性的原故。与磺酰胺类结合使用时,pentoxazone在移植水稻前一次施入田中具有良好的控制一年生和多年生杂草的能力。 Pentoxazone有效地控制抑制在叶绿素生物合成中的则叶啉-LX氧化酶。在光作用下,由于积累的原叶啉1X产生的活性氧使它诱导氧化物酶膜破裂。这种不同于其他除草剂的作用方式使用其成为控制ALS抗抑制剂的杂草,如母草屑dubia种的Majorpennell,母草属dubia类的dubiaPenell和久雨花属的Korsakowil等的重要工具。安全性 用于老鼠的毒性研究表明pentoxazone具有很低的毒性。在老鼠和细菌身上也不存在致畸和诱变可能性。在这些毒性研究的基础上,认为Pentoxazone是普通物质,基于动态毒性方面的研究,Pentoxazone对鱼、鸟和益虫存在很低或可忽略的毒性。 Pentoxazone对老鼠经口给药的大部分在168小时之内由类中排泄掉,少量被吸收的部分也迅速在肝脏中代谢掉和在粪便中排泄掉。在常规条件下应用于稻田时,pentoxazone很少转移至稻禾顶部。甚至在成熟期使用,它在稻子植物中也很快代谢掉,且在根、茎、叶的任何部位的残留量小于0.25ppm,特别在可食部分为0.046pmm。在有水时pentoxazone在土壤中的半衰期最高是40因,但它的活性成分和代谢物向下流动性很低,已查明对地下水系统没影响。因此,使用petoxazone对健康和环境具有深远的意义。
记者随机在北京新发地农产品批发市场、美廉美超市、昌平采摘园以及路边的草莓摊,购买8份草莓样品,送到北京农学院检测。经检测,样品全部检出农药乙草胺。乙草胺是一种除草剂,主要在大田作物里面,在草莓上不能使用,因而目前国家也没有登记草莓的残留标准。在美国,乙草胺已被列为b-2类致癌物,长期食用可能有致癌性。 乙草胺是一种广泛应用的除草剂。由美国孟山都公司于1971年开发成功,是目前世界上最重要的除草剂品种之一,也是目前我国使用量最大的除草剂之一。考虑到暴露在乙草胺每日摄取容许量以上对人体的潜在危害,以及地表水中乙草胺代谢物对人体的危害,现在还不能排除基因毒性的存在,欧盟委员会决定不予除草剂乙草胺再登记,已下令欧盟成员国在2012年7月23日取消其登记。现存库存的使用宽限期不能超过12个月。 乙草胺纯品为淡黄色液体,原药因含有杂质而呈现深红色。性质稳定,不易挥发和光解。不溶于水,易溶于有机溶剂。熔点大于0℃ ,蒸汽压大于133.3pa,沸点大于200℃,不易挥发和光解。30℃时与水的相对密度为1.11,在水中的溶解度微223mg/l。http://c.hiphotos.baidu.com/baike/s%3D220/sign=7c8d7269dab44aed5d4eb9e6831d876a/472309f790529822889b48e0d7ca7bcb0a46d46d.jpg 如此分子式,大家认为如何检测它呢?采用什么方法比较合适?包括前处理过程?是不是用氮磷检测器还是可以用液相?
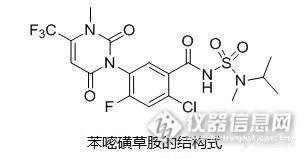
上世纪60年代。当时,杜邦公司开发出了首个脲嘧啶类除草剂—除草定,正式开启了该类除草剂研发的先河。而真正掀起脲嘧啶类除草剂开发热潮的是在上世纪90年代,当时人们对于该类除草剂的作用机理有了更深入的了解,发现脲嘧啶类除草剂属于原卟啉原氧化酶(PPO)抑制剂。杜邦公司在推出除草定后,又相继推出了异草定和特草定等产品。富美实的双苯嘧草酮以及先正达的氟丙嘧草酯均属于该类除草剂。而巴斯夫于2009年推出的苯嘧磺草胺(saflufenacil)更属于该类除草剂中的佼佼者。苯嘧磺草胺能够适用于多种生产系统和非耕地,在苗后或苗前均能使用;其次,适用作物多。苯嘧磺草胺能够用于包括谷物、玉米、棉花、水稻、高粱、大豆和果树等在内的30多种作物上;再次,防除谱广。苯嘧磺草胺能够防除90余种阔叶杂草,包括一些对三嗪类、草甘膦及乙酰乳酸合成酶抑制剂存在抗性的杂草。另外,它也具有作用快、残效期长等多种特性。http://ng1.17img.cn/bbsfiles/images/2017/02/201702010042_01_1623180_3.jpg2009年,苯嘧磺草胺在南美国家尼加拉瓜、智利和阿根廷三国登记。2010年,苯嘧磺草胺与精二甲吩草胺的复配制剂Verdict在美国获得登记,用于大豆。同年,苯嘧磺草胺正式登陆中国,以70%水分散粒剂(商品名:巴佰金)的形式面世,用于柑橘园和非耕地的杂草防除,由诺普信负责在中国市场的总经销。目前,苯嘧磺草胺已在美国、加拿大、中国、尼加拉瓜、智利、阿根廷、巴西和澳大利亚等国登记。苯嘧磺草胺可替代苯氧类除草剂2,4-D和磺酰脲类除草剂与草甘膦复配,可降低防治顽固性杂草对草甘膦的使用量。2014年,苯嘧磺草胺的全球销售额达到1.4亿美元。据巴斯夫公司预测,苯嘧磺草胺可实现3亿欧元的年峰值销售额。苯嘧磺草胺目前仍处于专利保护期中,其在中国的专利为巴斯夫于2001年申请的《尿嘧啶取代的苯基氨磺酰羧酰胺》,专利号为ZL01801896.3,对苯嘧磺草胺的化合物及合成方法进行了保护,该专利将于2021年4月30日到期.
丁草胺相关







 我要推广仪器
我要推广仪器
 下载APP
下载APP