分析二甲基丁醇,三甲基丁醇,样品浓度大约为99%。请教分析方法及使用何种色谱柱?谢谢了!![em58]
我用的极性柱测甲醇丁醇乙醇异丙醇只出三个峰,50度的程序升温,这是什么原因呢
[font=&][size=16px][color=#333131]可以做氨丁三醇(三羟甲基)氨基甲烷,杂质峰定性的检测机构请看过来[url=https://www.woyaoce.cn/helptest/detail-dcb6af847ef4628458ecdf6d407180cd.html]点击查看详情[/url][/color][/size][/font]
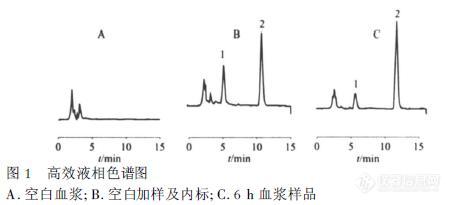
【作者】 马铭研; 周丹丹; 于治国;【机构】 沈阳药科大学药学院; 沈阳药科大学药学院 辽宁沈阳110016; 辽宁沈阳110016;【摘要】 目的:比较研究大鼠尾静脉注射与局部皮肤给予酮咯酸氨丁三醇的药动学行为。方法:采用HPLC法,色谱柱:Dia-monsil C18柱(200mm×4.6mm,5μm);流动相:甲醇-水-三乙胺-冰醋酸(80∶19.9∶0.02∶0.08);流速:1.0mL.min-1;柱温:30℃;检测波长:313nm。结果:酮咯酸氨丁三醇在0.2~100mg.L-1范围内与峰面积呈良好的线性关系(r=0.999 0),日内RSD为2.3%~5.1%,日间RSD为2.2%~12.2%,萃取回收率为86.8%~96.2%,注射剂和凝胶剂的T1/2α分别为(0.4±0.3)h,(2.9±2.6)h;T1/2β分别为(2.7±2.0)h,(9.0±8.5)h。结论:本试验建立的方法操作简单,方法灵敏、特异,结果准确。酮咯酸在大鼠体内药动学行为符合二房室模型;外用给药透皮吸收良好。【谱图】 http://ng1.17img.cn/bbsfiles/images/2012/08/201208142206_383898_1609970_3.jpg
三氧化二铝柱能不能进醇类?我想知道丁醇在三氧化二铝柱中的出峰位置,怎么实现?
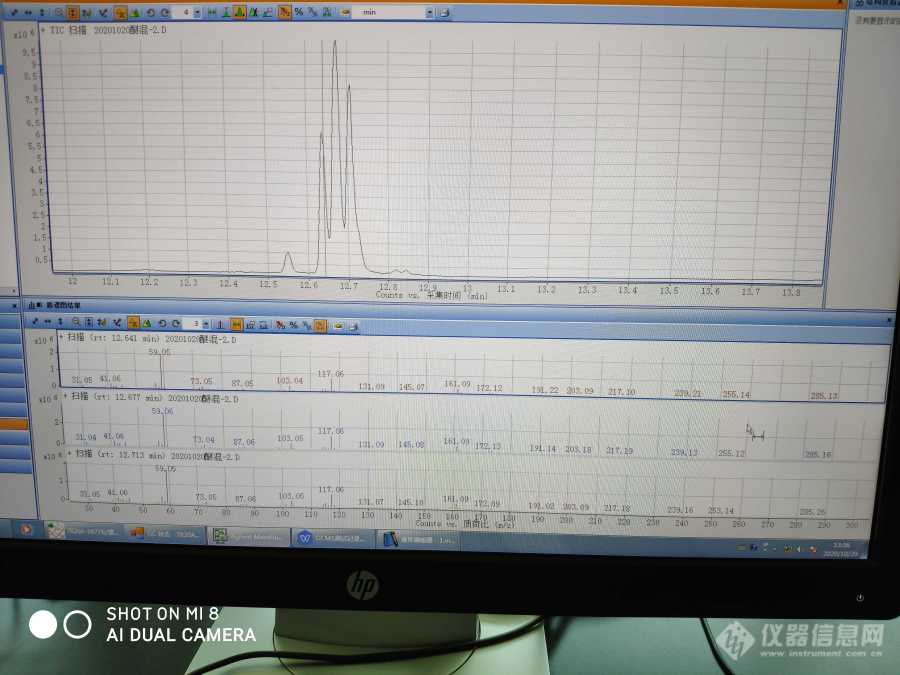
如题 柱子HP-5打出来的三丙二醇丁醚色谱图峰型这样跟手一样的 为什么会出现这种情况 柱温200[img]https://ng1.17img.cn/bbsfiles/images/2020/10/202010201337122955_4929_3960401_3.jpg[/img]
请问乙二醇、丙二醇、丁二醇和丙三醇混合物用什么型号的HPLC柱子效果好?
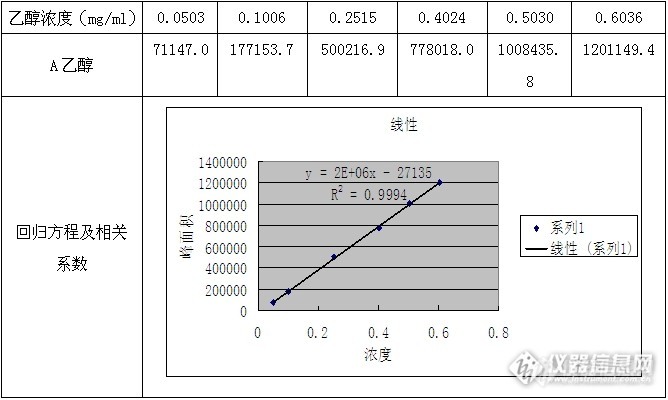
因工作需要,需要对酮咯酸氨丁三醇中的残留有机溶剂乙醇和1,2-二氯乙烷进行方法学研究,乙醇为三类溶剂,药典规定限度为0.5%,1,2-二氯乙烷为一类溶剂,药典规定限度为0.0005%,因为1,2-二氯乙烷的限度较低,在FID检测器下很难检测,故需要用到ECD检测器检测1,2-二氯乙烷。 方法学研究为,方法一,乙醇的检测;方法二,1,2-二氯乙烷的检测。1.1方法概述应用GC外标法对酮咯酸氨丁三醇中的残留有机溶剂乙醇进行定量分析。载气:氮气;检测器:FID。1.2对照品及样品名 称来源批号酮咯酸氨丁三醇样品某医药企业120201乙醇西陇化工股份有限公司11070111.3仪器和仪器参数气相色谱仪型号:岛津公司GC-2010天平型号:梅特勒公司XS105顶空进样器型号:DANI公司 HSS86.50色谱柱类型:DB-624 规格30m×0.53mm×3.0µm 载气:氮气 柱温:50 ℃检测器:FID检测器温度: 250℃;进样口温度: 200℃;流速: 3.0 ml/min;进样量: 1.0ml;分流比: 10:1样品盘平衡温度: 80℃;定量环温度: 90℃;传输线温度: 100℃;样品盘平衡时间: 30min1.4溶液配制对照溶液:准确称取乙醇50mg于100ml容量瓶中,用水稀释定容至刻度,摇匀,精密移取3ml置于20ml顶空瓶中,密封即得对照溶液。准确称取样品0.3g,置于20ml顶空瓶中,加水3.0ml,密封即得供试品溶液。1.5验证内容及结果1.5.1系统适用性试验方法:取酮咯酸氨丁三醇溶残对照溶液,依法连续进样5次,记录乙醇峰面积的相对标准偏差(RSD%)。乙醇峰面积的相对标准偏差RSD应不大于10%,乙醇的理论塔板应不小于10000,乙醇的拖尾因子应不大于1.5。结果:序号12345RSD%A乙醇[/si
请问何处可以购买色谱纯试剂: 磷酸三丁酯谢谢!
日前,我正在分析一种原料磷酸三丁酯的纯度,我查看的标准GBT 15354-2011 化学试剂 磷酸三丁酯,上面说用填充柱,但是目前手上没有这样的柱子,请问各位大侠有没有人做过这样的分析?推荐用什么色谱柱?谢谢
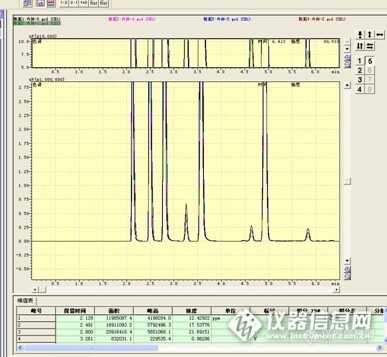
求助一下, ,我这个用程序升温法测甲醇,丙酮,正丙醇(内标) 丁醇 ,丁酯。 110℃ 2.5min 然后25℃/min升到160℃,并保持1min ,,为嘛每次都有三个小峰。如下图片
请教各位 样品是己酸乙酯 丙三醇 、2,3-丁二酮,要测定他们的重金属含量,怎样前处理 谢谢 ! 谢谢
我们用gcms顶空进样10微升的纯甲醇出现硼酸三甲酯无甲醇峰怎么回事?是与什么参加反应了吗?衬管?还是传输线毛细管柱?等等原因[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/03/201903161115414350_2763_2524334_3.png[/img]
離子源卸調後用甲醇洗後發現全氟三丁氨質譜圖中的770和866太低,高度接近峰底,遠遠沒有背景的119高,測試10溴deca出現上面相似 問題600以上峰都很低,不知道是離子遠電壓問題還是檢測器問題,燈絲也換過還是如此,求教大俠。!!
在用EDTA滴定溶液中的镁离子时在酸性条件下加入三乙醇胺做掩蔽剂,用铬黑T作指示剂,结果滴定的结果比未加入三乙醇胺时滴定的含量还高,这是什么原因导致的呢?恳请大家帮忙解释一下原因!
DMA做溶剂,却有醇峰,定位炔丁醇和混合溶剂跑出来的峰,混合溶剂里炔丁醇附近有两个峰,是反应了吗

http://ng1.17img.cn/bbsfiles/images/2015/03/201503301532_540105_1610895_3.jpg色谱条件:色谱柱: Diamonsil Plus C18-B 250*4.6 mm,5 μm(Cat#: 99409) 流动相: 0.3%(g/v)醋酸铵:甲醇=80:20 流速: 1 mL/min 柱温: 30 ℃ 检测器: UV 270 nm 进样量: 20 μL样品前处理: 精密量取本品适量,用流动相定量稀释制成每1 mL中约含氨丁三醇奥扎格雷56 μg的溶液。http://ng1.17img.cn/bbsfiles/images/2015/03/201503301551_540111_1610895_3.jpg
顶空GCMS中测试甲醇为啥一直产生硼酸三甲酯甲醇没有了
有证书的全氟三丁胺纯度99.7的可用作[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]调谐液吗
各位专家好,我想咨询一个问题,我的样品中含有乙二醇,丙三醇,丁四醇及其异构体部(在水溶液的体系中),请问各位专家,我应该采用什么方法来对其进行检测?需要哪些硬件设施?
三聚氰胺、瘦肉精(克伦特罗)、莱克多巴胺、沙丁胺醇胶体金快速检测卡,现在目前,有哪些家?
火焰光度检测器 S:8pg/s(十二烷硫醇) P:0.5pg/s(磷酸三丁酯) 这两个什么是意识?能告知一下吗
五种混标用二硫化碳溶解,毛细管柱为安捷伦DB1701,进样口温度250,检测器250,柱温35,程序升温至150,结果只检出甲醇,正丁醇,异戊醇。而乙二醇,异丙醇未检出,是我的仪器条件设置问题吗?求教!
小弟在做的溶剂标样:分别有甲醇,乙醇,异丙醇,乙酸乙酯,正丁醇,丁酮,甲苯,甲醚,甲基环乙烷,正丙酯,丁酯。。做标样的时候是这11种,但是出峰只出来9个,没有正丁醇和甲醚。。。然后单独进样的时候,正丁醇,正丙酯,甲醚的出峰时间很接近,用三种混的时候进样,却只有一个峰。不明白为什么进了三种只出了一个峰,是分离不开还是什么问题???柱温是70度。。检测器进样器都180度,毛细管柱50*25*25。。请各位大神帮忙..怎么分辨两种物质性质很相近?比如正丁醇,正丙酯,甲醚这三种为什么会那么近?
维权声明:本文为lunanjituan原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。酮咯酸氨丁三醇残留溶剂方法学验证(FID)一、方法:甲醇、乙醇、二氯甲烷、乙酸乙酯、乙酸、甲苯 称取异辛烷约20mg,精密称定,置50ml量瓶中,加DMF溶解并稀释至刻度,摇匀,作为内标贮备液;依次称取甲醇约300mg、乙醇约500mg、二氯甲烷约60mg、乙酸乙酯约500mg、乙酸约500mg、甲苯约89mg,均精密称定,置于50ml量瓶中,加DMF溶解并稀释至刻度,摇匀,精密量取溶液5ml,置50ml量瓶中,加DMF溶解并稀释至刻度,摇匀,作为对照贮备液;称取酮咯酸氨丁三醇约1.0 g,精密称定,置10ml量瓶中,加DMF适量使其溶解,加1.0 ml内标贮备液,加DMF稀释至刻度,摇匀,作为供试品溶液;精密量取对照贮备液5.0 ml,置10ml量瓶中,加1.0ml内标贮备液,用DMF稀释至刻度,摇匀,作为对照溶液。照气相色谱法(中国药典2005年版二部附录V E)和有机溶剂残留量测定法(中国药典2005年版二部附录VIII P第一法),用DB-624毛细管色谱柱(30m×0.53mm×3.0µm ),FID检测器,载气为氮气,进样口温度200ºC ,流速1.5ml/min,分流比5:1;柱温在40ºC下保留5分钟,然后以10ºC/min的速率升至200ºC,至少保留9分钟;检测器温度250ºC。分别精密量取上述两种溶液各1.0µl进样分析,记录色谱图,按内标法以峰面积计算,含甲醇、乙醇、二氯甲烷、乙酸乙酯、乙酸及甲苯分别不得过0.3%、0.5%、0.06%、0.5%、0.5%、0.089%。二、仪器与试药 岛津GC-2010气相色谱仪,FID检测器。甲醇(分析纯,国药集团化学试剂有限公司,批号:T20080109)、无水乙醇(分析纯,汕头西陇化工厂有限公司,批号: 0808141)、二氯甲烷(分析纯,汕头西陇化工厂有限公司,批号:0710311)、乙酸乙酯(沈阳瑞丰精细化学品有限公司,批号:20050626)、乙酸(上海试剂一厂,批号:20071201)、甲苯(天津瑞金特化学品有限公司,批号:20060704)、N,N-二甲基甲酰胺(分析纯,天津四友生物医学技术有限公司,批号:070914101)、异辛烷(分析纯,TEDIA company INC.USA,批号:506173)。三、色谱条件 色谱柱:DB-624毛细管气相色谱柱(30m×0.53mm×3.0µm )。FID检测器,载气为氮气,进样口温度200ºC ,流速1.5ml/min,进样量:1.0µl,分流比5:1;柱温:在40ºC下保留5分钟,然后以10ºC/min的速率升至200ºC,至少保持9分钟;检测器温度250ºC。尾吹:30ml/min。各峰的理论塔板数均应不低于10000,各峰的拖尾因子均应不大于2.0,各峰间的分离度均应不小于2.0。四、气相色谱方法学验证 照有机溶剂残留量测定法测定。根据合成路线,对甲醇、乙醇、二氯甲烷、乙酸乙酯、乙酸及甲苯等残留溶剂进行了方法学研究。1、专属性试验1.1空白基线 依柱温设定条件,得基线GC色谱图。见附图。1.2空白溶剂 精密量取DMF1.0µl 注入GC,得空白溶剂GC色谱图。见附图。1.3甲醇 称取甲醇约15mg,置于50ml量瓶中,加DMF溶解并稀释至刻度,摇匀。精密量取1.0µl 注入GC,得甲醇GC色谱图。见附图。1.4乙醇 称取乙醇约25mg,置于50ml量瓶中,加DMF溶解并稀释至刻度,摇匀。精密量取1.0µl 注入GC,得乙醇GC色谱图。见附图。1.5二氯甲烷 称取二氯甲烷约30mg,置于50ml量瓶中,加DMF溶解并稀释至刻度,摇匀,量取溶液1ml置10ml量瓶中,加DMF稀释至刻度,摇匀。精密量取1.0µl 注入GC,得二氯甲烷GC色谱图。见附图。1.6乙酸乙酯 称取乙酸乙酯约25mg,置于50ml量瓶中,加DMF溶解并稀释至刻度,摇匀。精密量取1.0µl 注入GC,得乙酸乙酯GC色谱图。见附图。1.7乙酸 称取乙酸约25mg,置于50ml量瓶中,加DMF溶解并稀释至刻度,摇匀。精密量取1.0µl 注入GC,得乙酸GC色谱图。见附图。1.8甲苯 称取甲苯约45mg,置于50ml量瓶中,加DMF溶解并稀释至刻度,摇匀,量取溶液1ml置10ml量瓶中,加DMF稀释至刻度,摇匀。精密量取1.0µl 注入GC,得甲苯GC色谱图。见附图。1.8异辛烷(内标) 称取异辛烷约20mg,置于50ml量瓶中,加DMF溶解并稀释至刻度,摇匀。精密量取1.0µl 注入GC,得异辛烷GC色谱图。见附图。1.9 对照溶液 依法配制对照溶液,精密量取1.0µl 注入GC,得对照溶液GC色谱图。见附图。由色谱图可知:待测各残留溶剂间的分离度均大于2.0,相互之间没有干扰。1.10 对照与样品混合溶液 依法配制对照溶液和样品溶液,并将二者按1:1相互混合,摇匀。精密量取1.0µl 注入GC,得对照与样品混合溶液GC色谱图。见附图。由色谱图可知:测定样品溶液中的潜在杂质对待测残留溶剂样品无干扰。2、系统适用性试验精密量取对照贮备液5.0ml,置10ml量瓶中,加1.0ml内标贮备液,加DMF稀释至刻度,摇匀,作为对照溶液。取对照溶液,依法连续进样六次,记录色谱图,记录各残留溶剂的保留时间、拖尾因子、理论塔板数、峰面积,计算各溶剂与内标峰面积的比值及RSD。结果见表1~7。http://ng1.17img.cn/bbsfiles/images/2010/12/201012091040_265708_2961690_3.jpghttp://ng1.17img.cn/bbsfiles/images/2010/12/201012091041_265709_2961690_3.jpg[img
水做溶剂,8ug/mL磷酸三丁酯标品,自动进样,峰面积10,RSD在10左右,太大了!做其他醇类样品RSD可以接受,换柱子和样品做磷酸三丁酯就不行,求做过的大神指点!
今天做β受体激动剂扩项,仪器是Waters Tq-s,流动相0.2%的甲酸水和乙腈,之前的标样是三天前配的,甲醇溶的标样,用30%乙腈稀释成梯度标液,配好的时候仪器软件出了问题,今天刚修好。进的时候沙丁胺醇响应很低,但西马特罗、克伦特罗、沙丁胺醇、莱克多巴胺、克伦特罗-D9、沙丁胺醇-D3都分开了,因为时间过了三天,所以曲线不是很好,今天拿母液用10%乙腈稀释的,可是突然发现西马特罗、沙丁胺醇和沙丁胺醇-D3三个分不开了。进了三个的单标,发现西马特罗没问题,但是沙丁胺醇和沙丁胺醇D3的峰还是出了双峰。不知道是哪里出了问题。求大神指点!
利用顶空[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定甲乙丙丁戊5种醇,分别测试了下正戊烷、正庚烷、丙酮、乙酸乙酯都能够与这5种醇分开,乙酸乙酯我已经选来做稀释剂,其他三种合适做内标物质吗?
1三氯丙醇国标18782中用的柱子是自制的,可以用哪种商品柱代替呢?是否有不需要过柱子的提取办法?2衍生用的七氟丁酰咪唑,需要加50uL,但是在网上查到的都是固体。标准里也没有提到这个东西是要配多大浓度。怎么办?
用TCD 检测三甘醇酯需要的担体固定液及色谱设定的条件







 我要推广仪器
我要推广仪器
 下载APP
下载APP