三七:三七归属五加科, 主根灰褐疙瘩形, 支根茎基也入药, 断面木部花纹显,饮片灰白蜡光泽, 理气止血散淤痛。 防己:防己归属粉防己,屈曲不直有结节,断面灰白车轮纹, 质坚而重不易折,饮片白色粉性大,祛风利水除湿热。 白芍:白芍来源毛茛科,圆柱条匀两头齐,纹理放射质坚实, 自来作用补血散, 柔肝止痛功效奇, 头痛眩晕止盗汗。 大黄:蓼科植物掌大黄,圆柱片块见锦纹, 槟榔查口特鲜明,紫外灯下棕荧光,气味特异有粘性,宣泻实热独有功。 银柴胡:石竹科有银柴胡, 珍珠盘头根头部,砂眼断处粉尘出,断面花纹特明显, 清热凉血疗骨蒸,阴虚发热清虚火。 白芷:白芷来源伞形科, 表面灰黄疙瘩丁,断面类白有粉性,气香浓烈辛微苦,形体类方杭白芷,散风祛湿排脓痛。 甘草:甘草药材属豆科,根茎灰棕或棕色, 胀果甘草根木质,光果甘草皮孔细,补脾益气又清热,调和诸药起功效。 柴胡:狭叶柴胡伞形科,根头膨大北柴胡,质韧难断气味香,根部较细南柴胡,质软易断败油气,升阳解表治感冒。 白薇:白薇来源萝藦科, 根茎粗短簇生根, 断面黄白木部黄, 毛大丁草习用品,茎粗灰棕长绵毛,清热凉血能利尿。 黄芪:豆科植物有黄芪,灰淡棕色质较韧,豆腥气味略微甘,断面菊花纹理状,补气固表排毒疮,心悸气短气血虚。 川牛膝:苋科植物川牛膝,根头膨大纵皱纹,黄棕灰褐质坚韧,切面三八同心环,活血祛风把湿利,利尿通淋关节痛。 白术:菊科植物有白术,拳状灰棕质坚硬,生晒白术显油性,断面花纹多油点,淡黄角质为烘术,健脾燥湿除痰饮。 白前:白前来源萝藦科,柳叶白前断中空,簇生细根织成团,根粗芫花叶白前,枝少稍弯不成团,化痰止咳气逆喘。 半夏:天南星科找半夏,光泽透明姜半夏,淡黄质松法半夏,上圆下尖水半夏,生品有毒可外用,化痰止呕治痈肿。 紫草:新蒙紫草紫草科,新疆紫草鳞层层,剥落碎片条长细,顶端残茎蒙紫草,墨汁臭味微苦涩,凉血活血治便秘。 南沙参:桔梗植物南沙参,顶端单双根茎显,断续环纹及纵沟,体轻质泡味微甘,饮片黄白多裂隙,养阴润肺虚热清。 天冬:百合科中天门冬, 黄白纺锤两顶尖,对光可见细中柱,断面角质半透明,质柔黏性甜微苦,化痰清热可滋阴。 麦冬:麦冬来源百合科,形似纺锤半透明,断面黄白质柔韧,味甜微苦有黏性,常用品种山麦冬,止咳润肺能滋阴。 北沙参:伞形植物珊瑚菜,干燥根茎北沙参,黄白颜色细长条,细直纹理显粗糙,皮木易分气特异,清肺泻火养阴好。 川贝母:贝母来源百合科,松贝个小怀抱月,青贝观音合掌状,炉贝马芽顶开裂,质硬而脆富粉性,润肺化痰能止咳。[font=Tahoma, 'Mic
实验室新人前来学习报道,各位大神多多关照
2020版《中国药典》变化,请指正https://simg.instrument.com.cn/bbs/images/default/em09503.gif 品名类别修订情况项目15版药典20版药典变化对照品,对照药材、试剂耗材、设备阿胶中药材修订鉴别、含量【鉴别】供试品离子流色谱中,应同时呈现与对照药材色谱保留时间一致的色谱峰。 【含量测定】本品按干燥品计算,含L-羟脯氨酸不得少于8.0%,甘氨酸不得少于18.0%,丙氨酸不得少于7.0%,L-脯氨酸不得少于10.0%。【鉴别】照【含量测定】特征多肽项下色谱、质谱条件试验。 【含量测定】氨基酸 本品按干燥品计算,含 L-羟脯氨酸不得少于8.0%,甘氨酸不得少于18.0% ,丙氨酸不得少于7.0% ,L-脯氨酸不得少于10.0%。 特征多肽 照高效液相色谱-质谱法(通则0512和通则 0431)测定。本品按干燥品计算,含特征多肽以驴源多肽A1(C41 H68N12O13)和驴源多肽A2(C51 H82 N18O18)的总量计应不得少于0.15%。 修订【鉴别】项条件同含量测定、【含量测定】“氨基酸”无变化,增加“特征多肽”驴源多肽A1(缺)、驴源多肽A2(缺)液相串联三重四极杆质谱仪阿胶饮片增订鉴别、含量【含量测定】.同药材。增加阿胶性状、 修订检査项,修订阿胶珠总灰分描述【含量测定】(氨基酸)同药材。即【含量测定】无变化,无需检测特征多肽液相串联三重四极杆质谱仪艾叶中药材修订含量【含量测定】本品按干燥品计算,含桉油精(C10H8O)不得少于0.050%。【含量测定】本品按干燥品计算,含核油精(C10H8O)不得少于 0.050%,含龙脑(C10H8O)不得少于0.020%。修订【含量测定】,方法改变,增加指标龙脑龙脑(已有)巴豆饮片增订鉴别/增加生巴豆性状/巴戟天饮片增订检查/增加饮片盐巴戟天【检查】 总灰分 同药材,不得过8.0%。增订总灰分标准下降白矾中药材修订鉴别、检查【来源】、【检查】铵盐 与氯化铵溶液混合液比较,不得更深。修订药材【来源】(描述增加)、【检查】 铵盐 取本品约0.lg,精密称定,照氮测定法(通则0704第二法或第三法,无需消解)测定,含铵盐以总氮(N)计,不得过0.3%。【检查】 铵盐标准提高白果饮片增订鉴别/增加白果仁和炒白果仁性状/白矶饮片增订鉴别/增加枯矶性状/白及中药材修订鉴别、含量药材【性状】【含量测定】/修订药材【性状】(描述增加)【含量测定】本品按干燥品计算,含1,4-二-2-异丁基苹果酸酯(C34H46O17)不得少于2.0%。增加【含量测定】1,4-二-2-异丁基苹果酸酯(缺)白及饮片增订鉴别、含量【性状】修订性状(增订描述)修订性状、增加含量测定【含量测定】同药材,含1,4-二[4-(葡萄糖氧)苯基卜2-异丁基苹果酸酯(C34H46O17)不得少于1.5%。增加含量测定1,4-二[4-(葡萄糖氧)苯基卜2-.异丁基苹果酸酯白前饮片修订检查/修订白前、蜜白前性状白前水分不得过12.0%(通则0832第二法)。 蜜白前水分不得过11.0%(通则0832第二法)水分标准提高白屈菜中药材修订鉴别【鉴别】显微鉴别/修订显微鉴别(描述删减)/白屈菜饮片增订鉴别/增加性状,删除其他检/白芍中药材修订重金属【重金属】铅不得过5mg/kg;镉不得过0.3mg/kg;砷不得过2mg/kg;汞不得过0.2mg/kg;铜不得过20mg/kg【重金属】铅不得过5mg/kg;镉不得过lmg/kg;砷不得过2mg/kg; 汞不得过0.2mg/kg;铜不得过20mg/kg。【重金属】镉标准下降,日常检测均合格白芍饮片修订鉴别/修订白芍饮片性状/白薇饮片修订鉴别/修订性状,无他项/白薇饮片增订鉴别、检查性状无描述、检查、浸出物无规定【性状】.本品呈不规则的段。根茎不规则形,可见圆形 凹陷的茎痕,结节处残存多数簇生的根。根细,直径小于 0.2cm,表面棕黄色。切面皮部类白色或黄白色,木部较皮部 窄小,黄色。质脆。气微,味微苦、【检查】(除杂质外)【浸出物】.同药材水分..不得过11.0%(通则0832第二法)。 总灰分..不得过13.0%(通则2302)。 酸不溶性灰分..不得过4.0%(通则2302)。 【浸出物】照醇溶性浸出物测定法(通则2201)项下的热浸法测定,用稀乙醇作溶剂,不得少于19.0%。 白芷中药材增订重金属【重金属】/【重金属】铅不得过5mg/kg;镉不得过lmg/kg;砷不得过2mg/kg; 汞不得过0.2mg/kg;铜不得过20mg/kg。增加【重金属】百部饮片增订检查/百部 【检查】 水分 不得过12.0%(通则0832第二法)。 蜜百部 【检查】 水分 不得过12.0%(通则0832第二法)。水分标准提高百合饮片增订检查/增加蜜百合性状、本品形如百合,表面棕黄色,偶见焦斑,略带黏 性。味甜。 【检查】(水分)同药材。无影响柏子仁饮片增订检查/ 柏子仁 除去杂质和残留的种皮。 【检查】 水分 不得过6.0%(通则0832第二法)。水分标准提高斑蝥中药材增订鉴别增加显微鉴别/
前些天请药店代煎的一副中药,药液密封在真空袋子里的,我家没有冰箱,就在阴凉处放到了第三天,昨晚喝的时候发现里边有咸味,就像是味精放多了。细看发现剩下的袋子里有气泡,白色浑浊和一些沉淀出现真奇怪,按说我的药材里没有含盐多的东西啊?按说密封的袋子里边的细菌啥的该是厌氧发酵的,喝了这种变质的中药会不会很危险啊。。里边会有哪些微生物?千万别有些出血性大肠杆菌啥的危险东西吧。真奇怪按理说煮了1个小时的中药里边的细菌不是应该被杀死了么?怎么还会繁殖?附上药方:嗽(肺经风寒)荆芥穗10g燀苦杏仁10g白前10g陈皮10g法半夏10g浙贝母10g子寇10g百部10g桔梗10g炙甘草5g炒紫苏子10g枇杷叶10g
做了一个12个物质的混标,发现部分酚类化合物曲线不呈线性,部分线性还可以。浓度梯度从2.5-200空白前后都做了一次,不存在问题诡异的结果,求指导
实验小白前来请教各位,液相的谱图是这样的话,处理的时候要不要手动定义基线,再积分?[img]https://ng1.17img.cn/bbsfiles/images/2019/03/201903172117582315_1408_3871770_3.png[/img]
今天做荧光,用的是和前几天一样的酸,一样的水,一样的瓶,一样的硼氢化钾空白前几天是70 200 左右空白今天突然变成 600 400然后还不断上升,清洗后会降低点,不过还是无法平衡,总是不断上升。新手啊,感觉好晕~
新人小白前不久做了一批样,但回收率特别不正常,想问问大神们,可能的问题出在哪里,还有仪器最近一段时间真空浮动范围很大,该怎么检查
按照国标GB/T 5009.145-2003方法做蔬菜农残检测,做按照标准方法进行空白前处理后,进行GC-MS检测,得到的图谱中出现十八烷等高分子烷烃的峰,我想可能是前处理是被溶解的凡士林的峰,不知道大家怎么理解的。
[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测铁为什么走曲线最后一个点的吸光度偏低接近空白,但空白前几个点吸光度正常,走了几次曲线都是这样 求助各位大神及同行
小白前来请教,主要是预实验用,血浆样品。配制了混标的Qc样品,浓度包括定量下限,LQC,MQC,HQC,不知道之后是取哪个浓度的Qc样品,加入多少μL内标液,内标液的浓度又是怎么确定,然后进行涡旋混匀、离心、取上清液进质谱。我看到的文献是这样的:制备含40、80、800、16000 ng/ml的Qc,取10μL,加入300μL内标(10 ng/mL),取200μL上清液进样品瓶。不知道这里面的原则是怎样的呢?
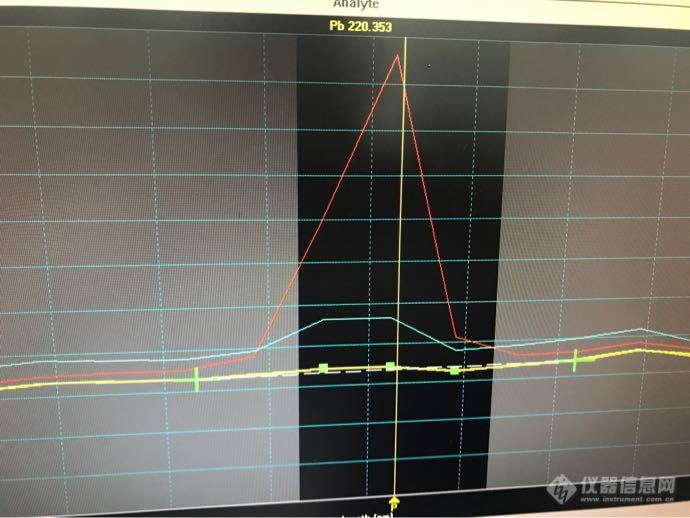
有没有ICP 大神在,问下仪器在走样分析的时候,标液的图谱也算正常,出的三角峰,但是在走样品的时候就会出现平峰,这种情况是什么原因造成的,有没有啥好的解决办法。谢谢。职场小白前来请教![img=,690,517]https://ng1.17img.cn/bbsfiles/images/2018/11/201811011357267654_3695_3348239_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2018/11/201811011358109151_4402_3348239_3.png[/img]
香精样品进样大家一般是直接进样还是处理过后进样呢?遇到粘度大的样品,大家是怎么解决的?大家通常用什么方法进行香精成分定量呢?小白前来提问,先谢谢老师们相助[img]https://simg.instrument.com.cn/bbs/images/default/em09502.gif[/img]
在造纸行业中,高岭土主要作为填料和涂料生产铜版纸和涂布白版纸。黏浓度是涂布造纸重要指标,一般在66%~70%之间。 高岭土粒级的分布对于矿浆的流变性有显著影响,直接影响着高岭土黏度。漂白前后4个粒级提纯样均显示出黏浓度随着粒径增大而递减趋势。黏浓度的影响因素很多,但归根到底在于高岭石本身固有的性质(内因)和外在条件(外因)。内因在于高岭土的成因、高岭石的晶体结构有序度、晶体形态以及粒度和粒度分布等;外因则在于矿物组成、分散剂种类含量、酸碱度以及加工条件、方法等。 研究表明,高岭石从高有序向无序转变,主要是沿着b轴方向出现了b/3的层位移,原始缺陷是由八面体空位的无序位移引起的;高岭石的结构无序化和晶体内部的缺陷有关,随着缺陷增加,晶体形态的均匀性受到破坏,晶体厚度变薄,径/厚比增大,边缘和角引起摩擦和破损,产生部分电荷不平衡,黏度增大。随着高岭石晶体的增大,晶体可发生卷曲,晶体结构发生位错的几率增高,不对称性增大,从而导致结晶度的降低。因此体现在提纯样品上,粒级越小,径/厚比越小,结晶度越高,单体含量越多,黏浓度越大;反之粒级越大,径/厚比越大,结晶度越低,集合体含量越多,黏浓度越小。 结论:通过提纯实验,获得0~15μm的4个粒级提纯样,所得高岭石百分含量高(97%、91%、80%和75%)、相应粒级分布百分比高(99.5%、68.1%、63.7%和72.9%),得到漂白前的黏浓度为63.99%、62.17%、58.49%和58.80%及漂白后的68.92%、63.98%、59.99%和59.29%。 2.茂名高岭石结晶有序度高、颗粒粒度细、径/厚比小、粒级分布均匀、杂质含量少是决定高黏浓度的关键因素,以样品a最为适合,其他粒级可通过相应技术改进,提高黏浓度。 3.保险粉漂白有利于黏浓度的提高,漂白前后黏浓度的差值与白度的差值高度正相关(R2=0.934);样品a到样品d,漂白效果逐渐降低,这可能与样品中炭质、有机质含量有关。 4.从样品a到样品d,随高岭石粒度分布范围增大,径/厚比增大,结构有序度降低,结晶度降低,与黏浓度呈高度正相关。同时,黏浓度与提纯样高岭石含量正相关,与杂质矿物石英、伊利石呈负相关性。
样品空白,随同样品处理时不添加任何样品但加入了样品处理的各个试剂,它能有效地避免样品前处理过程中带来的误差,因此是相当重要的。在进行原子荧光测定时,样品的测定值是扣除了样品空白后的值,因此,样品空白值的高低直接关系到样品值的准确性。一般在进行原子荧光测定时,应用标准空白观察仪器的稳定性,待仪器稳定后则进行相应的测定,但不知道大家对于样品空白的稳定性有没有相关认识。我的小经验:在进行样品空白测定时应反复对样品空白进行测定,观察样品空白前后测定间的稳定性,若前后测定值波动过大,则应反复测定几次,待前后测定值相对稳定时再进行样品的测定,这样可以避免因样品空白的波动造成的不准确性。
有没有做过甘草浸膏的,测甘草苷的对照品,为什么甘草苷后面出杂峰哪[img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808221438425145_6044_3461163_3.jpeg[/img]
问题一:什么是汞的记忆效应?问题二:先说我碰到的现象。用PE公司的ICP-OES,型号2100DV,测定汞。做完工作曲线后(曲线相关系数0.9995),用纯净水冲洗管路和雾化室,直到Hg(分析线194.168nm)的光强度到685,此时,把试剂空白(为6ml浓HNO3+2ml浓H2O2,稀释到50ml)当成未知样品进样,此时,光强度为69832,以为是空白受污染,用纯净水冲洗到光强度743时,不打试剂空白开始做样品。做完样品后,再一次把试剂空白当成未知样品测定,此时光强度为887。问什么试剂空白前后两次打出来的光强度相差那么大?
因为铬胶囊的问题,最近单位把几年前统一采购但是没有用的PE400安装好。在检测样品的过程中,发现两个问题,请教专家:1、每次测标准空白第一针,都会数值偏大,基本是第二针的两倍,导致RSD很大,我在测样品空白前空烧石墨炉,额外清洗针头都做了,但是还是会出现第一针数值大。2、报告打出来的数据中,样品的空白校正信号按理应该是扣除试剂空白,可实际上全部都扣除的是标准空白。就连试剂空白的校正信号也是扣除了标准空白。我找来找去,都找不到哪里设定可以改。抓狂!请教,这是因为软件的问题还是设定上哪里错误?标准空白不是指应该在标准样品中扣减么?怎么扣减到样品来了,试剂空白形同虚设啊。
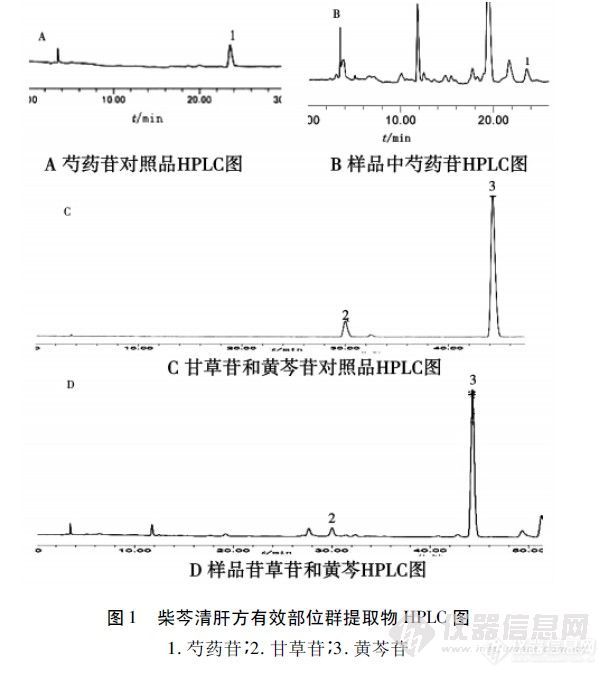
【作者】 付克; 张坤; 闫广利; 张春玲; 陈旭;【机构】 黑龙江中医药大学中医药研究院; 黑龙江中医药大学;【摘要】 目的:建立HPLC测定柴芩清肝方有效部位群提取物中黄芩苷、甘草苷、芍药苷的含量测定方法。方法:色谱柱:Diamonsil C18(4.6mm×250mm,5μm);流动相:乙腈-0.1%磷酸水溶液,梯度洗脱;检测波长为278nm(黄芩苷,甘草苷),230nm(芍药苷)。结果:黄芩苷、甘草苷、芍药苷分别在0.216~1.080μg(r=0.999 1)、0.020~0.100μg(r=0.999 5)、0.015~0.003μg(r=0.999 3)范围内线性良好,平均回收率分别为黄芩苷96.28%、RSD=0.83%(n=5);甘草苷97.2%、RSD=0.39%(n=5)和芍药苷98.1%、RSD=0.21%(n=5)。结论:该方法简便、快速、准确,具有良好的重复性和回收率,可作为柴芩清肝方有效部位群提取物的质量控制方法。 更多还原【关键词】 高效液相色谱法; 黄芩苷; 甘草苷; 芍药苷; 【基金】 黑龙江省科技攻关项目资助(GC08C322)http://ng1.17img.cn/bbsfiles/images/2012/08/201208061738_381991_2352694_3.jpg
三萜皂苷与甾体皂苷在薄层色谱上如何能分开?(甾体皂苷含量低于三萜皂苷)本人用硫酸乙醇显色出现的点都是紫红色的,没有墨绿色,并且表面看上去点都是分开的,会不会是甾体皂苷显示的颜色被三萜皂苷的盖住了。忘高人指点一下为谢!
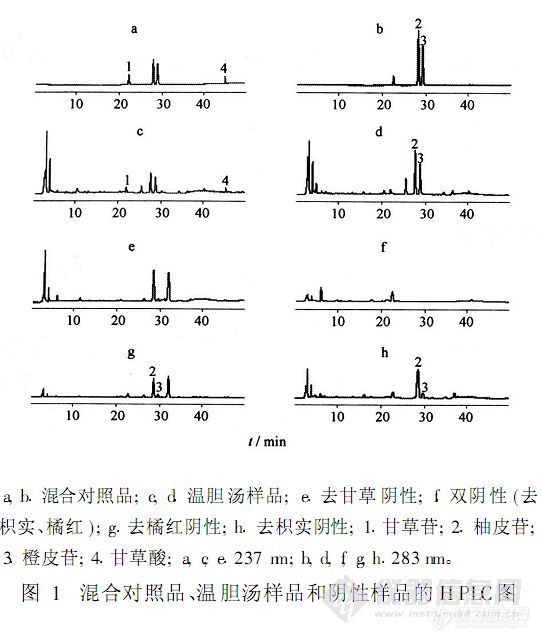
【作者】 许栋明; 程可建;【Author】 XU Dongming,CHENG Kejian(1.Science and Technology Innovation of Small and Mid-sized Enterprise Fund Management Center,Ministry of Science and Technology,Beijing 10038,China;2.Bescholor Research Center,Peking University,Beijing 100084,China)【机构】 科技部科技型中小企业技术创新基金管理中心; 北大世佳研究中心;【摘要】 目的:建立HPLC同时测定温胆汤中甘草苷、柚皮苷、橙皮苷和甘草酸含量的方法。方法:DIKMA Diamonsil(2)-C18柱(4.6 mm×250 mm,5μm);流动相乙腈(A)-0.1%磷酸溶液(B),线性梯度洗脱;检测波长237,283 nm;柱温25℃;流速1.0 mL.min-1;进样量10μL。结果:甘草苷、柚皮苷、橙皮苷和甘草酸铵的进样量与峰面积,分别在0.019 9~0.119(r=0.999 7),0.180~1.08(r=0.999 7),0.146~0.873(r=0.999 8),0.0393~0.236μg(r=0.999 7)呈良好的线性关系;平均加样回收率依次为97.7%,97.7%,97.1%,98.5%,RSD 1.4%,2.0%,2.0%,1.9%。结论:该方法快速,简便,重复性好,适合于同时测定温胆汤样品中甘草苷、柚皮苷、橙皮苷和甘草酸的含量。 更多还原http://ng1.17img.cn/bbsfiles/images/2012/08/201208131329_383466_2379123_3.jpg
我想做的是三磷酸腺苷二磷酸腺苷和磷酸腺苷,应该用什么柱子合适?
分析一中药山楂叶中金丝桃苷,出峰总是不好,总有一成分分不开,分离度不好的时候直接就叠成一个峰。因为山楂叶中还含有槲皮素(金丝桃苷的苷元也是槲皮素),请问分不开的这个峰是不是可能是槲皮素,怎么来检验呢?
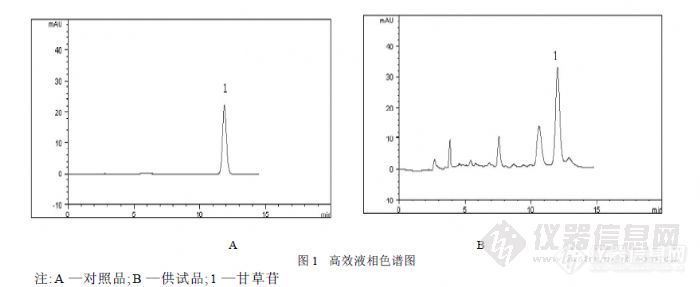
【作者中文名】 徐芳辉; 王强; 【作者单位】 湖南省益阳医学高等专科学校; 【摘要】 目的考察不同厂家甘草配方颗粒中甘草苷的含量。方法采用高效液相色谱法。色柱:DiamonsilC18柱(4.6mm×250mm,5μm);流动相:乙腈-1%冰醋酸(20∶80,V/V);流速:1.0mL/min;检测波长:320nm;柱温:30℃,进样量为10μL。结果甘草苷在148~2960ng范围内线性关系良好(r=0.9996),平均回收率为98.12%(RSD=1.21,n=5)。不同厂家甘草配方颗粒中甘草苷的含量为0.47~3.93mg/g。结论不同厂家产品中甘草苷的含量差异显著。【摘要】目的考察不同厂家甘草配方颗粒中甘草苷的含量。方法采用高效液相色谱法。色柱:DiamonsilC18柱(4.6mm×250mm,5μm);流动相:乙腈-1%冰醋酸(20∶80,V/V);流速:1.0mL/min;检测波长:320nm;柱温:30℃,进样量为10μL。结果甘草苷在148~2960ng范围内线性关系良好(r=0.9996),平均回收率为98.12%(RSD=1.21,n=5)。不同厂家甘草配方颗粒中甘草苷的含量为0.47~3.93mg/g。结论不同厂家产品中甘草苷的含量差异显著。http://ng1.17img.cn/bbsfiles/images/2012/08/201208011229_381006_1761902_3.jpg
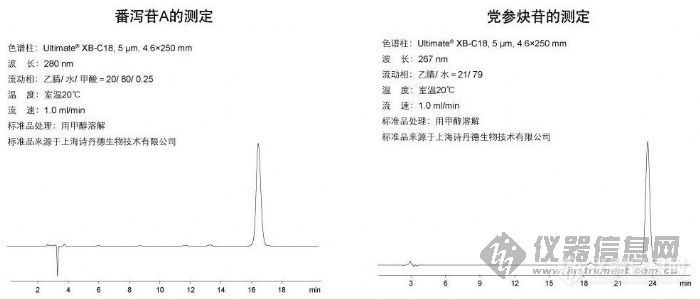
番泻苷A的测定和党参炔苷的测定http://ng1.17img.cn/bbsfiles/images/2009/10/200910161212_175963_1896702_3.jpg
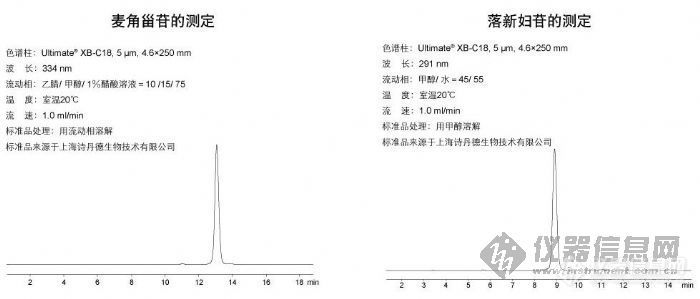
麦角甾苷的测定和落新妇苷的测定http://ng1.17img.cn/bbsfiles/images/2009/10/200910161207_175958_1896702_3.jpg

黄芩苷的测定和番泻苷B的测定http://ng1.17img.cn/bbsfiles/images/2009/10/200910161209_175959_1896702_3.jpg
漂白、增白后的纱线放置一定时间后,一是产生黄斑,二是随时间延长白度下降。 分析原因,其产生黄斑是由于漂白用水质不良,含二价铁离子较高所致,二价铁离子日久被氧化或三价铁离子,这样就在纱线上形成黄斑。这种黄斑不但影响外观质量,同时还会使纤维脆损。克服的办法是:遇水质不良时,纱线漂白前用草酸1.5-2.0g/L、在80-85℃下进行酸洗。因草酸能与铁离子生成络合离子而溶于水,水中的铁质即可去除,达到不泛黄的目的。 漂白、增白纱线随时间延长而白度下降的主要原因是由于煮练不充分。若纱线煮练不充分,棉纤维中杂质未完全去除,用这种纱线立即进行漂白、增白,时间一长,纤维内部的杂质及天然色素就会显露出来造成白度下降。克服的办法是:要保证漂白纱的毛效达到13-15cm/30min,而且毛效要均匀一致,才能保证漂白、增白纱线不泛黄。另外,用次氯酸钠漂白后脱氯时,大苏打用量过多或没有洗净,亦能使纱线泛黄。纱线烘燥时,烘房温度过高,烘燥时间过长也会造成纱线泛黄。
漂白后的纱线泛黄原因分析 漂白、增白后的纱线放置一定时间后,一是产生黄斑,二是随时间延长白度下降。 分析原因,其产生黄斑是由于漂白用水质不良,含二价铁离子较高所致,二价铁离子日久被氧化或三价铁离子,这样就在纱线上形成黄斑。这种黄斑不但影响外观质量,同时还会使纤维脆损。 克服的办法是:遇水质不良时,纱线漂白前用草酸1.5-2.0g/L、在80-85℃下进行酸洗。因草酸能与铁离子生成络合离子而溶于水,水中的铁质即可去除,达到不泛黄的目的。 漂白、增白纱线随时间延长而白度下降的主要原因是由于煮练不充分。若纱线煮练不充分,棉纤维中杂质未完全去除,用这种纱线立即进行漂白、增白,时间一长,纤维内部的杂质及天然色素就会显露出来造成白度下降。 克服的办法是:要保证漂白纱的毛效达到13-15cm/30min,而且毛效要均匀一致,才能保证漂白、增白纱线不泛黄。 另外,用次氯酸钠漂白后脱氯时,大苏打用量过多或没有洗净,亦能使纱线泛黄。纱线烘燥时,烘房温度过高,烘燥时间过长也会造成纱线泛黄。
各位高手,新人求助:我是刚使用原子吸收,是火焰法,没有石墨炉的,使用过程中出现以下问题1:标液,标液都是用5%硝酸配的,是不是标液都要用硝酸配,这样在测量标液之前的调零是吸水还是吸硝酸?2:标液用硝酸稀释后,被测样品是不是也要用硝酸稀释?3:在样品空白前的“样品调零”有的时候会调不到零,是要按“调零”强制调零还是吸光度可以不为零?有时样品调零后吸光度为负值,导致测得浓度为负值?4:在测量过程中吸光度数值很不稳定,是什么因素导致的,一般吸光度在什么范围内变动可视为稳定?5:测量后一定要先关乙炔气体,火焰自动熄灭吗?可以点熄火键吗,但是会产生蜂鸣声,为什么?6:测量的数据保存后还能不能修改?这个数据只有原子吸收的软件能打开,还有其他的软件能打开吗?7:每个元素是不是都对应一个最佳的测量浓度范围,一般标液要配到什么浓度,还是每个元素都不一样?







 我要推广仪器
我要推广仪器
 下载APP
下载APP