【题名】:饮用水中有机磷酸酯的健康风险及双酚A和未知消毒副产物识别【全文链接】: https://cdmd.cnki.com.cn/Article/CDMD-10056-1022681999.htm
我用的是化学纯的双酚A ,perkin serries200的高效液相检测,以甲醇:水=7:3为流动相出现肩峰,后来用磷酸调样品PH=3,结果出现三个峰,为什么啊?
可否有达人参照2010新版药典二部,完成了双嘧达莫片含量测定项目?鄙人在做这个项目的时候发现其流动相存在问题,双嘧达莫物质峰不出现,怀疑是被强保留在色谱柱上了。具体分析原因为:双嘧达莫有关物质项中采用流动相为:[font=宋体]磷酸氢二钠溶液[/font][font='Times New Roman','serif'][[/font][font=宋体]取磷酸氢二钠[/font][font='Times New Roman','serif']250mg[/font][font=宋体],加水[/font][font='Times New Roman','serif']250ml[/font][font=宋体],溶解后,滴加磷酸溶液[/font][font='Times New Roman','serif'](1→3)[/font][font=宋体]调节[/font][font='Times New Roman','serif']pH[/font][font=宋体]值至[/font][font='Times New Roman','serif']4.6]-[/font][font=宋体]甲醇[/font][font='Times New Roman','serif']=([color=#ff483f]25:75[/color]), 而双嘧达莫片含量测定采用流动相为:[font=宋体]磷酸氢二钠溶液[/font][font='Times New Roman','serif'][[/font][font=宋体]取磷酸氢二钠[/font][font='Times New Roman','serif']1.0g[/font][font=宋体],加水[/font][font='Times New Roman','serif']1000ml[/font][font=宋体],溶解后,滴加磷酸溶液[/font][font='Times New Roman','serif'](1→3)[/font][font=宋体]调节[/font][font='Times New Roman','serif']pH[/font][font=宋体]值至[/font][font='Times New Roman','serif']4.6]-[/font][font=宋体]甲醇[/font][font='Times New Roman','serif']=([color=#fe2419]75:25[/color]),这两个检测项目所用流动相,差别是相当的大。不知道是否属于刊误呢?ps:2005版药典中,双嘧达莫片含量测定采用滴定法。[/font][/font]

10,抽取5个版友);中奖名单:玲儿响叮当(注册ID:jshbhh)mengzhaocheng(注册ID:mengzhaocheng)捌道巴拉巴巴巴(注册ID:v3082413)千层峰(注册ID:jxyan)dahua1981(注册ID:dahua1981)http://ng1.17img.cn/bbsfiles/images/2016/09/201609271558_612317_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/09/201609271558_612318_708_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================双嘧达莫片方法:HPLC基质:药品应用编号:101392化合物:双嘧达莫固定相:Platisil ODS色谱柱/前处理小柱:Platisil ODS 5u 150 x 4.6 mm样品前处理:【含量测定】 取本品20片,除去包以后,精密称定,研细,精密称取适量(约相当于双密达莫50mg),置100ml量瓶中,加水10ml,超声15min,加甲醇75ml,振摇30min,用甲醇稀释至刻度,摇匀,滤过,精密量取续滤液2ml,置25ml量瓶中,用流动相稀释至刻度,摇匀。另取双密达莫对照品适量,精密称定,加流动相溶解并稀释制成每1ml中含双密达莫40ug的溶液。色谱条件:检测波长:UV 288 nm 流动相:磷酸氢二钠溶液-甲醇(75:25) 磷酸氢二钠溶液:取磷酸氢二钠1.0g,加水1000ml溶解后,滴加磷酸溶液(1→3)调节pH至4.6。 洗脱方式:等度 进样量:20 ul文章出处:P76关键字:双嘧达莫片,2010版中国药典,HPLC,含量测定,铂金,Platisil ODS谱图:http://www.dikma.com.cn/Public/Uploads/images/shuangmi.GIF图例:1. 双嘧达莫
我今天色谱峰很怪,第一个物质和第三个物质都出双头峰,双头峰的峰高和峰面积都差不多,而第二个物质和其余的物质都是一个很对称的峰,我实在不知道什么回事?我的前三个物质都中有手性碳原子。有没有老师也遇到这样的情况啊?怎么解决啊?我的柱子是Agilent C8 的,流动相是14%甲醇,2%THF,84%30mM磷酸缓冲溶液,pH7.2.谢谢了!等等回音
高效液相色谱 SB_C18柱 流动相~磷酸缓冲盐.乙腈 测布洛芬与双氯芬酸 这两个峰分不开 我应该怎么调流动相比例 或者怎么调梯度 或者流速 梯度与分峰之间的规律是什么 有大神能给普及一下么
请帮忙分析原因我用HPLC法分析1,6-二氢-2-甲基-6氧代[3,4‘-双吡啶]-5-甲腈时,分别用过C8,C18色谱柱,流动相用的是5mmol/l的磷酸氢二钠(pH=7.0)与乙腈甲醇以一定比例混合,使用过程中,柱压也没有升高的现象,供试品及对照品溶液的pH值分别为4.5和7.2,但色谱柱的柱效下降很快。后来我又尝试着调节pH值分别为6.5、5.8、4.0、均没有很大的起色,用这根色谱柱分析别的化合物峰形很差,难道真是这种化合物对色谱柱有损伤?两根色谱柱连续用三天以后均会如此,为什么?请帮忙分析。谢谢!!
我在做液相时遇到了不会解决的问题,希望各位前辈能帮帮我。我在做利福平时(按照药典上的条件,流动相:甲醇-乙腈-0.075mol/L磷酸二氢铵-1.0mol/L枸橼酸)利福霉素的峰第一次拖尾,第二次就出现双头峰。之后就一直是双头峰!!!!其他的峰都很正常。(柱子是新的,用过两次。也排除了保护柱的影响)哪位能帮我分析一下!!!很急!!!!
我在做液相时遇到了不会解决的问题,希望各位前辈能帮帮我。我在做利福平时(按照药典上的条件,流动相:甲醇-乙腈-0.075mol/L磷酸二氢铵-1.0mol/L枸橼酸)利福霉素的峰第一次拖尾,第二次就出现双头峰。之后就一直是双头峰!!!!其他的峰都很正常。(柱子是新的,用过两次。也排除了保护柱的影响)哪位能帮我分析一下!!!很急!!!!
色谱CHINESE JOURNAL OF CHROMATOGRAPHY2005 Vol.23 No.1 P.12-17 高速逆流色谱双水相体系分离蛋白质Separation of Proteins in Aqueous Two-Phase Systems with High-Speed Counter-Current Chromatography郅文波 邓秋云 宋江楠 顾铭 欧阳藩 摘 要:利用多分离柱高速逆流色谱仪,研究了聚乙二醇1000(PEG1000)-磷酸盐双水相体系的固定相保留率及该体系对蛋白质混合物和鸡蛋清样品的分离.以14.0%PEG1000-16.0%磷酸盐体系的上相为固定相,在流速0.6mL/min和转速900 r/min的条件下,固定相的保留率达到33.3%.在pH 9.2的PEG1000-磷酸盐双水相体系中,细胞色素C、溶菌酶和血红蛋白的分配系数差异最大,采用该pH值的14.0%PEG1000-16.0%磷酸钾盐双水相体系,在流速1.0 mL/min和转速850 r/min的条件下,成功地分离了这3种蛋白质的混合物.鸡蛋清中的主要蛋白质成分--卵转铁蛋白、卵白蛋白和溶菌酶在pH 9.2的15.0%PEG1000-17.0%磷酸钾盐体系中也具有最大的分配系数差异.采用该体系,在流速1.0 mL/min和转速850 r/min的条件下,成功地分离了鸡蛋清样品,得到的卵白蛋白、溶菌酶和卵转铁蛋白的电泳纯度分别为100%,100%和60%,收率均大于90%.关键词:高速逆流色谱 双水相体系 蛋白质 分离纯化分类号:O658 文献标识码:A文章编号:1000-8713(2005)01-0012-06 作者简介:郅文波,男,博士研究生,E-mail:zhiwenbo@163.com.通讯联系人:欧阳藩,男,教授,Tel:(010)82627061,Fax:(010)62561822,E-mail:fouyang@home.ipe.ac.cn. 作者单位:郅文波(中国科学院过程工程研究所生化工程国家重点实验室,北京,100080) 邓秋云(上海同田生化技术有限公司,上海,200122) 宋江楠(中国科学院过程工程研究所生化工程国家重点实验室,北京,100080) 顾铭(中国科学院过程工程研究所生化工程国家重点实验室,北京,100080) 欧阳藩(中国科学院过程工程研究所生化工程国家重点实验室,北京,100080) [img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=23113]高速逆流色谱双水相体系分离蛋白质[/url]
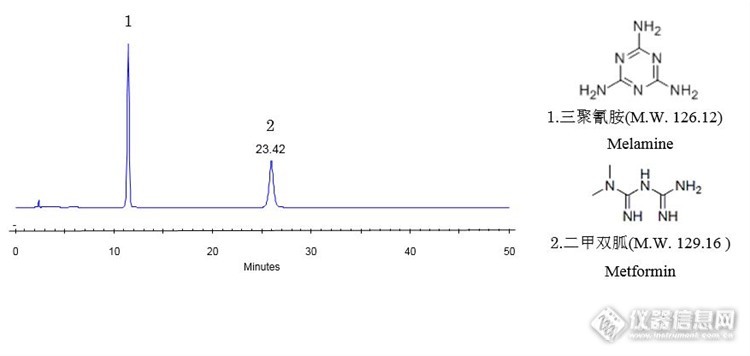
在9月29日的帖子中,我们介绍了在强阳离子交换模式下,使用CAPCELL PAK SCX UG80色谱柱对双胍类化合物进行保留与分离,并且尝试缩短分析时间的实验分析例。(详见 http://bbs.instrument.com.cn/topic/6289670)今天将要为大家带来的是使用该色谱柱,按照2015年版《中国药典》方法对盐酸二甲双胍进行的分析数据。盐酸二甲双胍(Metformin HCL)为白色结晶性粉末,无臭。在水中易溶,在甲醇中溶解,在乙醇中微溶,在氯仿或乙醚中不溶。熔点为220~225℃。分子式:C4H12ClN5分子量:165.6246以下为使用资生堂强阳离子交换色谱柱CAPCELL PAK SCX UG80对盐酸二甲双胍检测得到的谱图,请参考。http://ng1.17img.cn/bbsfiles/images/2016/10/201610271024_615216_2222981_3.jpg色谱柱:CAPCELL PAK SCX UG80 ;4.6mm i.d.×250mm流动相:1.7%W/V磷酸二氢铵(磷酸调pH至 3.0)流 速:1.0mL/min温 度:40 °C检 测:PDA 218nm进样量:20 µL样 品:5 µg/mL(按照药典方法配置)*注:峰上标数字为分离度。
O2SI公司推出纺织品中禁限用磷酸酯类阻燃剂混标(6组分) CDGG-110893-01-1ml 1000mg/L于乙腈,1 ml各组分成分如下:双(2,3-二溴丙基)磷酸酯 Bis(2,3-dibromopropyl) phosphate DDBPP 5412-25-9磷酸三(β-氯乙基)酯 Tris(2-chloroethyl)phosphate TCEP 115-96-8磷酸三(2-氯丙基)酯Tris(2-chloropropyl)phoaphate TCPP 13674-84-5三-(1,3-二氯异丙基)磷酸酯 Tris-(1,3-dichloropropyl)phosphate TDCP 13674-87-8三(2,3-二溴丙基)磷酸酯 Tri-(2,3-dibromopropyl)-phosphate TRIS 126-72-7
求教大神,血浆稀释的替考拉宁标准品,流动相是乙腈和磷酸盐,岛津C18的柱子,现在有的样品杂志峰出现双头峰,而且杂质峰高逐渐增加,样品定量也不准确,但是同一批中也有正常的杂质峰,正常的峰形中样品定量准确,这是什么原因?请教大家
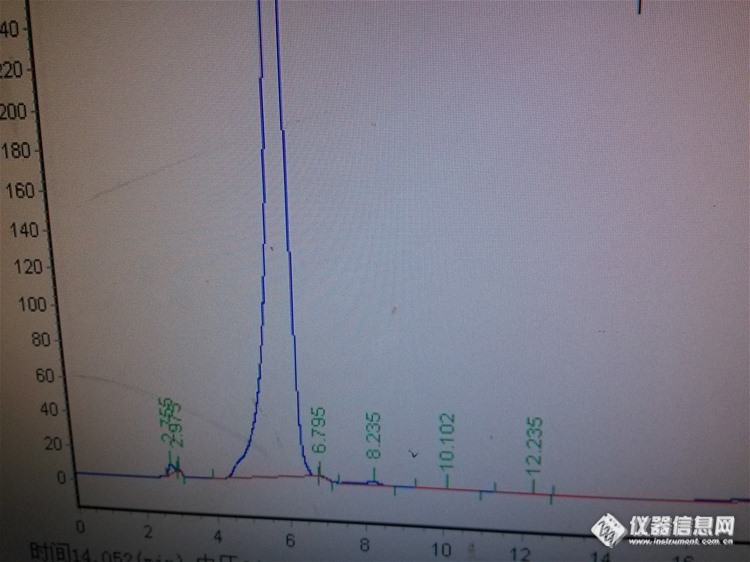
岛津LC16 双泵液相程序 0-10min 40%乙腈 10-20min40-70%乙腈 20-30min70%乙腈 30-40min70-40%乙腈水相是磷酸二氢钾 用三乙胺 和磷酸调节PH=3.1周一开机发现柱子(从2月22日开始 用这个条件分析一直用这个柱子)压力报警 于是就更换了同一型号的 岛津柱子峰型包裹了前面的杂质峰http://ng1.17img.cn/bbsfiles/images/2017/10/2016031713595543_01_2328726_3.jpg于是就把 原来的柱子再生后使用结果程序到32min 出现一个大包http://ng1.17img.cn/bbsfiles/images/2017/01/201701191700_667625_2328726_3.jpg现在怎么处理才能把这个鬼峰去掉呢? 已经进行的处理有 :更换柱子 跟换保护住 清洗整个流路(异丙醇) 水相的水用娃哈哈纯净水 瓶装 以前一直用桶装水都没有效果今天把水相换成纯水 走了一遍 程序 发没有这个峰了 换回水相 有出现是不是这个缓冲盐有问题?
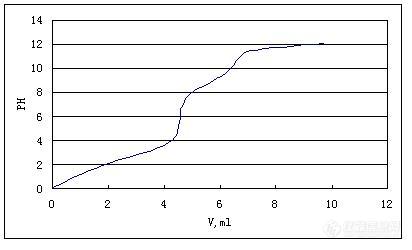
前不久接到顾客的一个投诉,说是用我们的有机产品进行磷酸化后,生产出的磷酸酯外观看起来比较稀,润湿力差。自己查过自己的留样也没有什么异常。于是便考虑测一下磷酸酯里的单双酯的含量。不过磷酸酯毕竟不是我们的产品,也没测过。便做了一些摸索性实验。实验过程虽然不是一帆风顺,但是正是前面的一个又一个的小失败,才指导我们最终走向成功,并且也让我越来越喜欢动手做实验。我们这有台电位滴定仪,加上磷酸本身有三个电离常数,K1=7×10-3, K2=6×10-8, K3=4×10-13. 所以决定用非水相电位滴定法。第一次做的时候,不知道里面单酯双酯磷酸各有多少,估摸着称了1克样,用0.1N的氢氧化钾进行滴定。样品称多了,氢氧化钾溶液用了40毫升还没滴完。所以第一次实验结果“失败”第二次,于是便减小了称样量,称了0.2克。加了30毫升乙醇作溶剂,将样品搅拌溶解后,还是用0.1N的氢氧化钾进行滴定。这回消耗的氢氧化钾溶液虽然不多了,但始终第三级突跃几乎看不到。又反复做了二次,滴定时该注意的都注意了,连滴定时用的杯了都反复洗了好几便,还是失败。于是找来相关的书仔细研究,发现原来是因为三级电离常数太小,很难直接滴定出来。[img]http://ng1.17img.cn/bbsfiles/images/2009/12/200912211104_191063_1932487_3.jpg[/img]第五次,有了前面的失败,本是胸有成竹的。同样称了0.2克。加了30毫升乙醇作溶剂,用0.1N的氢氧化钾进行滴定,当第二等当点结束后,PH为11时,按STOP, 暂时停止滴定。加了2毫升的氯化钙饱和溶液,这时PH下降了,然后再滴定。似乎有一点的起伏变化,但仍然不太明显。依然是失败。第六次,于是加了10毫升的氯化钙饱和溶液,这时PH下降更多,然后再滴定。终于看到了明显的突跃。[img]http://ng1.17img.cn/bbsfiles/images/2009/12/200912211105_191064_1932487_3.jpg[/img]计算就很简单了:单酯= (V2-V3)*CKOH*Mw单酯/m样品重量 双酯= (V2-V1)*CKOH*Mw单酯/m样品重量磷酸= V3*CKOH*Mw单酯/m样品重量经过前面多次的实验,终于成功了!一种说不出的兴奋与激动涌上心头。在实验的过程中,不断的失败,也不断的收获。让我也越来越喜欢DIY做实验。
逆流色谱应该属于液相色谱各位同行们,我现在开始专门高分离纯化方面的研究,目前主要着手用双水相体系分离蛋白质,现在遇到了问题,请教各位:我用的是PEG磷酸盐体系,我对比了国产的、俄罗斯的和sigma的PEG1000 分离效果差异很大,不知道你们做的结果如何?另外,进口的PEG价格比较贵,请问有什么办法回收重复使用?希望能够得到同行的指点,不胜感谢!我的Email:guanghui_9@163.com
色谱条件与系统适用性试验:用辛烷基硅烷键合硅胶为填充剂;以甲醇-水-三乙胺(70:30:0.3)(含庚烷磺酸钠10mmol/L,用磷酸调pH至4.0)为流动相;检测波长为260nm。理论板数按克霉唑峰计算不低于2500。 文献报道的方法: 刘晓琳等用RP-HPLC法测定双唑秦栓中甲硝唑、克霉唑和醋酸氯己定的含量。仪器:Agilent 1100高效液相色谱仪。色谱柱以辛烷基硅烷键合硅胶为填充剂;以甲醇-水-三乙胺(70:30:0.3)(含庚烷磺酸钠10mmol/L,用磷酸调pH至4.0)为流动相;检测波长为260nm;流速:1.0ml/min;柱温:25℃。 黄东萍等用RP-HPLC法测定双唑秦栓中甲硝唑、克霉唑和醋酸氯己定的含量。仪器:Waters高效液相色谱仪。色谱柱为Kromasil C18(150mm×4.6mm);以甲醇-水相(庚烷磺酸钠3g,三乙胺2.5ml,加水至1000ml,冰醋酸调pH至2.5)(65:35)为流动相;流速:1.0ml/min;检测波长:甲硝唑315nm,克霉唑和醋酸氯己定为260nm。 范荣等用RP-HPLC法测定双唑秦栓中甲硝唑、克霉唑和醋酸氯己定的含量。仪器:LC-10AT高效液相色谱仪。色谱柱为VPP-ODS柱;流动相:水-乙腈-冰醋酸(70:30:1);检测波长254nm;流速:1.0ml/min;柱温:室温。
色谱柱是强酸性阳离子交换基团键合全多孔不规则形硅胶固定相,即SCX柱,做的样品是盐酸二甲双胍,流动相是1.7%的磷酸二氢铵溶液。总是在进空白即流动相时候在10分钟左右出现一很大的大峰,换了不同的仪器、试剂和柱子还是有峰,请教各位有没有好的方法。
我们生产 阿莫西林 用液相检测中间反应,色谱柱12个小时都在用,运行样品时间总共3个小时左右,其他时间是10%甲醇水,流动是 磷酸氢二钾:乙腈=97.5:2.5 用过好多色谱柱,寿命都在一个月左右,双头峰 拖尾。用保护住 时间长了压力就很高以至于不能进样。 求教。
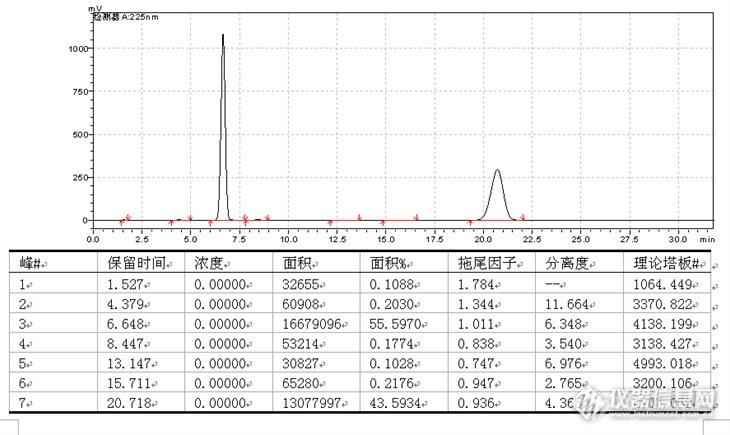
前言:这是一个老项目了,对于公开品名,是已经上报了,宝刀系列均是以前的资料,写出来和大家共享一下。由于网络问题,有些图看不到了,我在附件上显示了。建议大家看附件好了,看这个很吃力。望谅解。项目:含量测定(3.2.P.5.2.9)检查方法:照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定试验条件:仪器:LC-10AT VP(SHIMADZU) SPD-10A VP(SHIMADZU)万分之一电子天平(Sartorius ABS-124S型)工作站(LCsolutionlite色谱工作站)色谱柱(welchrom 填料:C18,规格:250mm×4.6mm,填料粒径:5μm;pn:wel518425,sn:w10212097)UV检测器(检测波长:225nm)柱温:室温流动相:流动相A为0.1mol/L磷酸二氢钾溶液-0.018mol/L十二烷基硫酸钠-甲醇-乙腈(275:275:200:250),用磷酸调节pH值至2.0;流动相B为乙腈。按下表进行线性梯度洗脱: 时间(分钟)流动相A(%)流动相B(%)090105851579010119010流速:1.2ml/min运行时间:约11分钟系统适用性:理论板数按阿莫西林峰和双氯西林峰计算应均不低于2000,双氯西林与阿莫西林的分离度应符合规定。具体试验操作:取装量差异项下的内容物,混合均匀,精密称取适量(约相当于阿莫西林25mg,双氯西林12.5mg),置100ml棕色量瓶中,加磷酸盐缓冲液(0.05mol/L磷酸二氢钾溶液-甲醇-乙腈(550:200:250)并稀释至刻度,摇匀,滤过,精密量取20μl注入液相色谱仪,记录色谱图;另取阿莫西林和双氯西林对照品,精密称定,加磷酸盐缓冲液(0.05mol/L磷酸二氢钾溶液-甲醇-乙腈(550:200:250)溶解并定量稀释制成每1ml中约含阿莫西林0.25mg和双氯西林0.125mg的溶液,同法测定,按外标法以峰面积分别计算出供试品中C16H19N3O5S和C19H17Cl2N3O5S的含量。计算公式:标示量百分含量(%)=××100%式中:Cs为对照品的浓度(mg/ml);At为供试液的主峰面积;Nt为供试液的稀释倍数;AS为对照品溶液的主峰面积;W为供试品取样量(mg)。3.2.P.5.3.6 含量测定色谱图见附件1367~1442含量测定方法学验证结果概要 项目验证结果波长选择[size=9pt
中文名称: 羟丙基二淀粉磷酸酯 中文商品名称:羟丙基磷酸双淀粉 英文名称: Hydroxypropyl distarch phosphate 别名: HPDSP 详情: 理化性质:白色粉末,无臭,无味,易溶于水,不溶于有机溶剂。在醚化的基础上,适当地交联所得到的HPDSP,其膨润力、透明度仍显著高于原淀粉。糊液对温度、酸度和剪切力的稳定性高。 来源与制法: 淀粉与三偏磷酸钠或磷酰氯(≤0.1%)与环氧丙烷(≤10%)伴同酯化而成。 编辑本段毒理学依据 1、ADI:无须规定(FAO/WHO,1994)。 2、可安全用于食品(FDA,§172.892,1994)。 质量要求:质量标准(FAO/WHO,1990;CXAS,1991) 羟丙基含量/% 7.0 氯丙醇/(mg/kg)≤ 1 土豆或小麦类淀粉/% ≤ 0.14 其他类淀粉/5 ≤ 0.04 二氧化硫 谷物类/(mg/kg)≤50 其他类/(mg/kg)≤10 砷(以As计)(mg/kg) ≤ 3 重金属(以Pb计)(mg/kg) ≤ 40 铅/(mg/kg)≤ 2 编辑本段用途与注意事项 我国《食品添加剂使用卫生标准》(GB2760―2007)表A.3(可在各类食品中按生产需要适量使用的添加剂名单)第46为羟丙基二淀粉磷酸酯,功能为增稠剂。未限定最高用量,可按需添加。 FAO/WHO规定:可单独使用或与其他增稠剂合用。用于蛋黄酱,5 FAO/WHO;罐装胡萝卜(产品含有奶油或其他油脂)、发酵后经加热处理的调味酸奶及其制品,10 g/kg;冷饮制品,30 g/kg;罐装沙丁鱼和沙丁鱼类产品,20 g/kg;罐装鲐鱼和竹荚鱼,60 g/kg(仅用于填料);速冻鱼条和鱼块(仅指用面包粉和面包拖料包裹),以GMP为限。羟丙基二淀粉磷酸酯Hydroxypropyl Distarch Phosphate编码 GB 20.016;INS 1442性状 白色粉末,无臭,无味,易溶于水,不溶于有机溶剂。在醚化的基础上,适当地交联所得到的HPDSP,其膨润力、透明度仍显著高于原淀粉。制法 由淀粉在碱性条件下,与环氧丙烷进行醚化,再与磷酸交联剂进行酯化反应制得。质量标准 参见羟丙基淀粉。鉴别方法 本品呈一般食变性淀粉反应和磷酸盐反应。1.一般食用变性淀粉反应 同羟丙基淀粉鉴别方法1、2、3。2.磷酸盐反应 参见磷酸三钙。毒理学依据1.GRAS FDA-21CF
各位大侠,聚磷酸盐/焦磷酸盐/三偏磷酸盐/六偏磷酸的离子色谱方法谁有做过没?能否给个参考,谢谢!
请问水中磷酸根、磷酸氢根、磷酸二氢根用AS11柱能分开么,什么条件?
流动相中常用到缓冲盐如磷酸二氢钾,磷酸氢二钾,磷酸二氢钠,磷酸氢二钠等,不知道这些缓冲盐有什么区别
磷酸化的Ser、Tyr和Thr修饰的多肽:我们国肽生物提供单磷酸化和多磷酸化多肽服务,目前我们已经能够提供四个磷酸化位点修饰的多肽。[img=,434,186]https://ng1.17img.cn/bbsfiles/images/2019/04/201904191649355554_2764_3531468_3.jpg!w434x186.jpg[/img][img=,486,498]https://ng1.17img.cn/bbsfiles/images/2019/04/201904191649358877_2957_3531468_3.jpg!w486x498.jpg[/img]我们主要提供:多肽定制、磷酸肽、生物素标记肽、荧光标记肽、同位素标记肽、人工胰岛素、药物肽、多肽合成、目录肽、偶联蛋白、化妆品肽、多肽文库构建、抗体服务、糖肽、订书肽、RGD环肽等。合肥国肽生物官网:http://www.bankpeptide.com欢迎咨询服务热线:17718122172;17718122684;17730030476;17718122397
大神,求个磷酸铁 磷酸铁锂 中 金属元素的测试标准。。。。
今天做实验,遇到有趣的事情,看看大家怎么看流动相:甲醇+水、甲醇+0.1%磷酸、甲醇+0.2%磷酸、甲醇+0.5%磷酸同一比例下,对物质的保留时间差异很大,如何选择,有诀窍吗?谈谈你的观点和认识http://simg.instrument.com.cn/bbs/images/default/em09511.gif
本来是离子色谱的新手,刚接手单位新来的赛默飞的ics-900,现在在做水质无机阴离子的测定的方法验证,请问一下里面所提到的磷酸氢根和磷酸根有什么区别,是不是一样的东西,是否可以代替,有没有磷酸氢根的国标贮备液买???
十二水合磷酸氢二钠好像比较容易风化,那怎么直观的证明十二水合磷酸氢二钠没有变质?大家实验室的十二水合磷酸氢二钠是不是直接使用,或是可以105摄氏度烘成无水磷酸氢二钠??
我想做的是三磷酸腺苷二磷酸腺苷和磷酸腺苷,应该用什么柱子合适?







 我要推广仪器
我要推广仪器
 下载APP
下载APP