http://simg.instrument.com.cn/bbs/images/brow/emyc1004.gif急求谁有YS/T710《氧化钴化学分析方法》---第1部分:钴量的测定 电位滴定法; ---第2部分:钠量的测定 火焰原子吸收光谱法; ---第3部分:硫量的测定 高频燃烧红外吸收法; ---第4部分:砷量的测定 原子荧光光谱法; ---第5部分:硅量的测定 钼蓝分光光度法; ---第6部分:钙、镉、铜、铁、镁、锰、镍、铅和锌量的测定 电感耦合等离子体发射光谱法。
BS 3482-9-1991 干燥剂试验方法.第9部分:氯化钴含量的测定
请问哪位前辈有[url=http://www.51zbz.com/biaozhun/54234.html]GB/T 9282.1-2008 透明液体 以铂-钴等级评定颜色 第1部分:目视法[/url]
我的56邮箱中的部分化学资料,主要是新药、天然药物化学资料,在hailiangxin@56.com邮箱共享区,今天刚到这个论坛,觉得很不错,希望给大家有所贡献,也希望加分鼓励。今后会发更多更好的帖子,谢谢了!
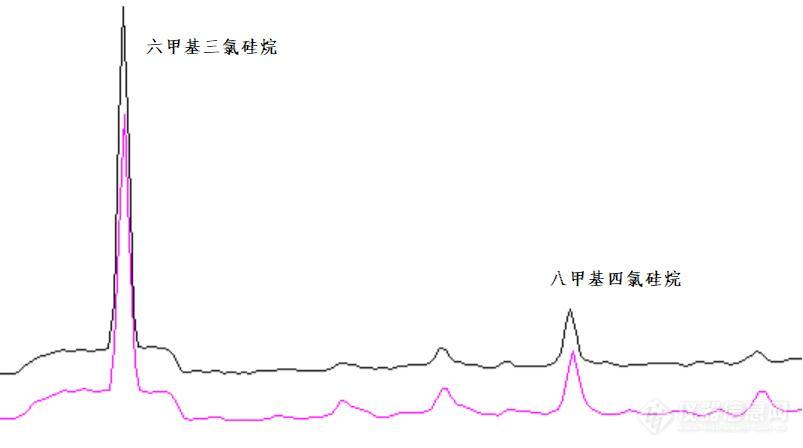
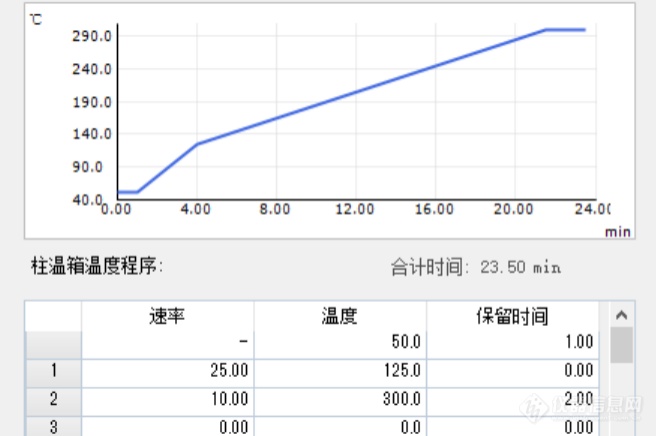
不分流进样条件下的溶剂问题之 - 固定相流失[align=center]概述[/align]不分流进样时,实际进入到色谱柱的溶剂量较大,可能会造成色谱柱固定相的流失程度增大。某些分析情况下,固定相的流失会影响分析结果。下文以某分析故障为例,予以说明。[align=center]故障现象[/align]某用户使用Shimadzu [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS-QP2020 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱联用仪,分析某医药包装材料中的多甲基环硅氧烷类的物质残留,实验结果出现某些组分的定量重复性不良。实验分析条件如下所示:仪器型号: [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS-QP2020[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url]进样口温度: 280℃色谱柱: Rtx-5MS 30m*0.25mm*0.25um程序升温: 50℃(1min)-20℃/min - 150℃(0min)-10℃/min -300℃(5min)进样量: 1ul,不分流进样检测器: 230℃,scan方式采集用户反映待测物质中的六甲基环三硅氧烷定量结果重复性较差,重复进样过程中峰面积RSD大于2%。[align=center]故障预判[/align][url=https://insevent.instrument.com.cn/t/Mp]gc[/url]或者[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS重复性不良的常见原因主要是系统存在泄漏、系统工作状况不稳定、系统安装状态不标准或者样品不良等原因。最常见的故障原因与进样口相关,准备好进样口的相关部件,赶往用户。[align=center]故障处理[/align]首先对用户的多个分析数据进行了考察,发现用户样品中参与定量的多个硅氧烷组分的重复性表现不同。六甲基环三硅氧烷目标峰的重复性较差,而八甲基四环硅氧烷、十二甲基环流硅氧烷等目标组分的重复性良好,如图1所示。既然不同的组分重复性表现不同,那么故障的原因并非来自硬件的共性问题,应当与样品自身性质或者样品的分析方法有关。此外,六甲基三氯硅烷的色谱峰形状也存在不良的问题,色谱峰的底部存在异常的基线扰动。[align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/09/202109200654054632_6002_1604036_3.jpg[/img][/align][align=center]图1 两次实验谱图比较[/align]检查色谱柱安装情况,石英棉填充状态,进样口的泄漏情况均未见异常。进样[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS出厂验收的标准样品,数据重复性结果良好,确认[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS系统硬件无异常。考虑到本分析案例中,目标物质为甲基环硅氧烷类,与色谱柱或者进样隔垫等耗材的流失特征峰一致。再次考察了[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS的程序升温空白,重复采集空白数据,比较所有数据中的柱流失峰,发现重现性良好。排除分析系统本底信号不稳定的问题。该谱图中六甲基三氯硅烷的色谱峰明显不良,色谱峰底部存在明显的平台状态干扰。借鉴顶空和VOC等弱保留物质分析的经验(弱保留组分分析时,一般需要避免使用不分流方式,否则容易产生峰形异常),将进样方式改为分流,连续进样多次,数据重复性良好,六甲基三氯硅氧烷的峰形状也得到了改善,峰强度同时降低。[align=center]原因剖析[/align]在不分流状态下,大体积溶剂的进样对固定相进行冲刷,使固定相的流失发生了不稳定的增加。但改用分流进样后,进入色谱柱的溶剂量显著减少,减弱了固定相的流失。[align=center]小结[/align]本案例较为特殊,溶剂会影响重复性,会冲刷色谱柱的固定相,如果分析方法的目标物为甲基硅氧烷类物质,可能会造成定量误差。不分流进样时,溶剂对色谱柱的清洗,会造成定量结果的不稳定。
样品进行RoHS测试时有不合格部分,进行WEEE中3R评估时如何考虑?是否不合格的部分不能计算为可回收?或者直接出产品不符合WEEE的要求?求高人指点!
JB/T 8941.2-2014 一般用途罗茨鼓风机 第2部分:性能试验方法
求助:国家鼓励发展的资源节约综合利用和环境保护技术(综合利用部分)
GB/T 23367.2-2009 钴酸锂化学分析方法第2部分:锂、镍、锰、镁、铝、铁、钠、钙和铜量的测定电感耦合等离子体原子发射光谱法》GB/T 23367.1-2009 钴酸锂化学分析方法第1部分:钴量的测定EDTA滴定法其他版本也可以
[font=&][size=16px][color=#191919]固体废物危险特性鉴别时能否只检测部分关键指标分为两种情况。情况一:如果选择检测的部分指标结果已经证实了该废物具有危险特性时且检测结果满足判断该鉴定对象的类别时,则可以不再检测其他指标。情况二:如果已经优先检测的指标未证实该废物具有危险特性,则通过前期分析认为有必要检测的其他指标仍需要继续检测。[/color][/size][/font]
27日,张家界武陵源、天门山、大峡谷景区等部分景区将恢复开放,A级景区向全国游客半价,黄龙洞对当地抗疫医护终身免票;
(恒温分析)式中:t’R为校正保留时间;Z和Z+1分别为目标化合物(X)流出前后的正构烷烃所含碳原子的数目;这里:t’R(z)CH2 993 CrH2 1284 CH- 65 CrH- 966 C -657 Cr 198 =CH2 1259 =CH- 133……含氧基团18 -OH 39719 1-OH 74720 2-OH 64521 3-OH 56123 -O- 71525 -O-OH 202羰基基团26 -HCO 60227 CO 52429 -CO-O- 51531 -CO-OH 1383含氮基团37 -NH2 511卤素基团60 -F -29含硫基团71 -SH 561含硅基团79 SiH- -308含磷基团82 P- 28883 PO- 28884 PS- 288可以看出在极性柱子上面gn值一般要大一些。一般讲在极性柱子上面化合物出峰要慢一些。基团对保留指数贡献的增加值(gn)也就大一些,特别是极性大一些的基团。*********************************1. 部分醇类估算举例(正构醇h取350值)正戊醇, CH3(CH2)4OHpolar: RI = 113*1 + 99*4 + 397*1 + 350 = 1256 (测定值:1253)(注:甲基-CH3的贡献增加值是113,亚甲基CH2为99,羟基-OH为397,参表2 )己醇:CH3(CH2)5OHNonpolar: RI =113+99*5+397+350=1355 (测定值:1355)庚醇Nonpolar:RI=113+99*6+397+350=1454(测定值:1465)辛醇Nonpolar: RI=1553(测定值:1550)壬醇Nonpolar: RI=1652(测定值:1663)癸醇Nonpolar: RI=1751(测定值:1769)十一醇Nonpolar: RI=1850(测定值:1866)十二醇Nonpolar: RI=1949(测定值:1965)Isoamyl alcohol:RI=113*2+99*2+6+747+40=1217(测定值:1212)2部分醛类估算举例(醛h取-15值)己醛 C6,CH3CH2CH2CH2CH2HCO:RI=113+99*4+602+(-15)=
YS/T 349.1-2009 硫化钴精矿化学分析方法 第1部分:钴量的测定 电位滴定法YS/T 349.2-2010 硫化钴精矿化学分析方法 第2部分 铜量的测定 火焰原子吸收光谱法YS/T 349.3-2010 硫化钴精矿化学分析方法 第3部分 锰量的测定 火焰原子吸收光谱法YS/T 349.4-2010 硫化钴精矿化学分析方法 第4部分 二氧化硅量的测定 氟硅酸钾容量法镉化学分析方法
如题,现单位用到乙酸铁、乙酸铜、乙酸钴原料,但目前没有其检测方法,不知哪位大侠提供一下,谢谢了
分享下:)2010版本的GB150[1].1-2010固定式压力容器 第1部分:通用要求(征求意见稿)
哪位朋友有标准GB 16915.1-2003 《标准家用和类似用途固定试电气装置的开关 第1部分:通用要求 》文本,谢谢!
今发现一氦气钢瓶上面的阀开的最大,不漏气,而开一部分(阀未全旋开)就有点漏气,请问是何缘故?
部分处理部分处理
请帮忙找一下以下资料,谢谢 1 IEC 68 基本环境试验规程 2 IEC 384-1:1982 电子设备用固定电容器 第一部分:总规范 3 GB 2693-90 4 GB/T 2471-1995 电阻器和电容器优先数系 5 GB/T 2691-94 电阻器和电容器的标志代码 6 IEC410(1973) 计数检查抽样方案和程序 7 IECQ/QC 001001(1986) IEC电子元器件质量评定体系(IECQ)基本章程 8 IECQ/QC 001002(1986) IEC电子元器件质量评定体系(IECQ)程序规划jenscwj@126.com
[font=&][size=18px]分流进样和不分流进样在操作参数的设置,对样品的要求以及衬管结构方面也有很大区别,下面分别讨论之.[/size][/font][font=&][size=18px]分流进样[/size][/font][font=&][size=18px](一)载气流路和衬管选择[/size][/font][font=&][size=18px]分流进样时载气流路如图4-2a所示.进入进样口的载气总流量由一个总流量阀控制,而后载气分成两部分:一是隔垫吹扫气(1~3mL/min),二是进入汽化室的载气.进入汽化室的载气与样品气体混合后又分为两部分:大部分经分流出口放空,小部分进样色谱柱.以总流量为104 m1/min为例,如果隔垫吹扫气流设置为3m1/min,则另101mL/min进入汽化室.当分流流量为100mL/min时.柱内流量为lml/min,这时分流比为100:1.注意.此仪器设计将柱前压调节阀置于分流气路上,这就可在总流量不变的情况下,改变柱前压.柱前压越高,柱流速越大,分析速度越快.而要在柱前压不变(柱流速不变)的条件下改变分流比,则必须调节总流最.总流量越大,分流比越大.[/size][/font][font=&][size=18px]分流进样口可采用多种衬管,用于分流进样的衬管大都不是直通的,管内有缩径处或者烧结板,或者有玻瑞珠,或者填充有玻璃毛.这主要是为了增大.与样品接触的比表面,保证样品完全汽化.减小分流歧视〔见下面关于分流歧视问题的讨论).同时也是为了防止固体颗粒和不挥发的样品组分进入色谱柱.注意,填充物应位于衬管的中间,即温度最高的地方,也是注射器针尖所到达的地方,这样对提高汽化效率,减少注射器针尖对样品的歧视更为有效.另外,玻璃毛活性较大,不适合于分析极性化合物.此时可用经硅烷化处理的石英玻璃毛.[/size][/font][font=&][size=18px]衬管的上端常用"O"形硅橡胶环密封.用一段时间后该环会老化而造成漏气.故要及时更换.当进样口温度超过400℃时,最好采用石墨密封环.[/size][/font][font=&][size=18px](二)样品的适用性[/size][/font][font=&][size=18px]分流进样适合于大部分可挥发样品,包括液体和气体样品,特别是对一些化学试剂(如将剂)的分折.因为其中一些组分会在主峰前流出.而且样品不能稀释,故分流进样住往是理想的选择.此外,在毛细管GC的方法开发过程中,如果对样品的组成不很清楚.也应首先采用分流进样口对于一些相对"脏"的样品,更应采用分流进样,因为分流进样时大部分样品被放空,只有一小部分样品进入色谱柱,这在很大程度上防止了柱污染.只是在分流进样不能满足分析要求时(灵敏度太低),才考虑其他进样方式,如不分流进样和柱上进_样等.[/size][/font][font=&][size=18px]总之,分流进样的适用范围宽,灵话性很大.分流比可调范围广,故成为毛细管GC的首选进样方式.[/size][/font][font=&][size=18px]不分流进样[/size][/font][font=&][size=18px](一) 载气流路和衬管选择[/size][/font][font=&][size=18px]不分流进样与分流进样采用同一个进样口,顾名思义,不分流进样就是将分流气路的电磁阀关闭[图4-2(b)],让样品全部进入色潜柱.这样做的好处是显而易见的,既可提高分析灵敏度,又能消除分流歧视的影响.然而,在实际工作中,不分流进样的应用远没有分流进样普遍,只是在分流进样不能满足分析要求时(主要是灵敏度要求),才考虑使用不分流进样.这是因为不分流进样的操作条件优化较为复杂.对操作技术的要求高.其中一个最突出的问题是样品初始谱带较宽(样品汽化后的体积相对于柱内载气流量太大).汽化的样品中溶剂是大量的,不可能瞬间进入色谱柱,结果溶剂峰就会严重拖尾,使早流出组分的峰被掩盖在溶剂拖尾峰中[如图4-3(a)所示],从而使分析变得困难,甚至不可能.有人也将这一现象叫做溶剂效应.[/size][/font][font=&][size=18px]消除这种溶剂效应可从几个方面考虑,但就载气的流路来说,主要是采用所谓瞬间不分流技术.即进样开始时关闭分流电磁阀,使系统处于不分流状态[图4-2(b)].待大部分汽化的样品进入色醉柱后,开启分流阀,使系统处于分流状态[图4-2(a)].这样,汽化室内残留的溶剂气体(当然包括一小部分样品组分)就很快从分流出口放空,从而在很大程度上消除了溶剂拖尾[如图4-2(b)所示].分流状态一直持续到分析结束,注射下一个样品时再关闭分流阀.所以我们说,不分流进样并不是绝对不分流,而是分流与不分流的结合.这里,确定一个瞬间不分流时间(从进样到开启分流阀的时间)往往是分析成败的关键.原则上讲,这一时间应足够长.以保证绝大部分样品进人色谱柱,避免分流歧视的影响 同时又要尽可能短,以最大限度地消除溶剂抢尾,使早流出峰的分析更为准确.这显然是有矛盾的.在实际工作中,常常是根据样品的具体情况(如溶剂沸点,待测组分沸点和浓度等)或操作条件来确定一个优化的折衷点.研究结果表明,这一时间值一般在30~80S之间.文献报道多采用0.75min,即从进样到开启分流阀的时问为0.75min,通常能保证95%以上的样品进入色谱柱,本节后而将介绍如何用实验方法确定优化的不分流时间.[/size][/font][font=&][size=18px]衬管的尺寸是影响不分流进样性能的另一个重要因素.为了使样品在汽化室尽可能少地稀释,从而减小初始谱带宽度,衬管的容积小一些有利,一般为0.25~1mL,且最好使用直通式衬管.当用自动进样器进样时,因进样速度快,样品挥发快,故建议采用容积稍大一些的直通式衬管.对于干净样品,衬管内可不填充玻璃毛,对于相对脏的样品,则需要填充玻瑞或石英毛,以保证分析的重现性并保护色谱柱不被污染.但要注意,由于不分流进样时样品在汽化室滞留的时间比分流进样时长,热不稳定化合物的分解可能性也大,故衬管和其中填充的石英毛都必须经硅烷化处理,且要及时清洗,更换和重新硅烷化.[/size][/font][font=&][size=18px](二)样品的适用性[/size][/font][font=&][size=18px]不分流进样具有明显高于分流进样的灵敏度,它通常用于环境分析(如水和大气中痕量污染物的检测),食品中的农药残留监测,以及临床和药物分析等.这些药品往往都比较脏,所以样品的预处理是保护色谱柱所必须注意的问题.此外,待测痕量组分如果在溶剂拖尾处出蜂,还可采用溶剂聚焦的方法来提高分析灵敏度.[/size][/font][font=&][size=18px]不分流进样对样品溶剂有较严格的要求.因为进样口温度,色谱柱初始温度,瞬间不分流的时间和进样体积都与溶剂沸点有关.一般地讲,使用高沸点溶剂比低沸点溶剂有利,因为溶剂沸点高时,容易实现溶剂聚焦,且可使用较高的色谱柱初始温度,还可降低注射器针尖歧视以及汽化室的压力突变.表4-2列出了常见的溶剂及其沸点和实现溶剂聚焦宜采用的色谱柱初始温度.[/size][/font][font=&][size=18px]另一方面,洛剂的极性一定要与样品的极性相匹配,且要保证溶剂在所有被测样品组分之前出峰,否则早流出的峰就会被溶剂的大峰掩盖.同时,溶剂还要与固定相匹配,才能实现有效的溶剂聚焦.必要时可采用保留间隙管来达到聚焦的目的.[/size][/font][font=&][size=18px]对于高沸点痕量组分的分析,不分流进样就容易多了.此时可以不考虑溶剂的沸点,因为有周定相聚焦就完全能保证窄的初始谱带,采用高的初始柱温还可缩短分析时间.事实上,不分流进样应是分析高沸点痕最组分的首选方法.[/size][/font]
“色”路蹒跚,萧规曹随,浅谈固体制剂溶出度方法学部分题外话:我们吃的不是含量,是生物利用度;溶出度技术是评价固体制剂的灵魂与核心所在。以上是谢沐风老师说的,很在理。定义:大多数口服固体制剂在给药后必须经吸收进入血液循环,达到一定血药浓度后方能奏效,从而药物从制剂内释放出并溶解于体液是被吸收的前提,这一过程在生物药剂学中称作溶出,而溶出的速度和程度称溶出度,从药品检验的角度上讲,溶出度系指药物从片剂或胶囊等固体制剂在规定的溶剂中溶出的速度和程度。 过去认为只有难溶性药物才有溶出度的问题,但近年来研究证明,易溶性药物也会因制剂的配方和工艺不同而致药物溶出度有很大差异,从而影响药物生物利用度和疗效,在USP中规定测定溶出度的制剂有相当数量是易溶性药物。质量研究中溶出度的内容:溶出度测试方法学和溶出行为方法学。溶出度研究试验主要包括以下内容:(1)溶出介质的选择,(2)溶出介质体积的选择,(3)溶出方法(转篮法与桨法)的选择,(4)转速的选择,(5)溶出度测定方法的验证,(6)溶出度均一性试验(批内),(7)重现性试验(批间)等。检验测试方法检验方法-方法学验证1,检测波长的确定(辅料以及胶囊的干扰,胶囊一是对测试干扰,而是对样品崩解的影响)2,空白试验3,滤膜干扰验证4,线性试验5,回收率6,溶液稳定性滤膜干扰试验:对照品用离心对比。线性试验(50/60% 限度 100%)回收率溶液稳定性(0.5h 1.0h 2.0h 8.0h)溶出度方法溶出方法考察1,溶出转速选择2,溶出介质选择3,溶出限度和取样时间的确定4,溶出条件的确定5,溶出均一性考察6,三批检验溶出介质/检测波长的选择:关键点为做曲线,取样时间点5、10、20(15)、30、45、60分钟。不同的介质可能影响到波长的选择,要和对照品一致。溶出介质体积的选择:漏槽试验,参照标准。一般是500/900/1000ml溶出装置的选择:利用标准方法进行试验,做曲线。一般是胶囊或易上浮样品用篮法,片一般用桨法。具体做法篮法[font=Times New Roma
[align=center][font=宋体][size=14.0000pt]不分流进样条件下[/size][/font][font=宋体][size=14.0000pt]的[/size][/font][font=宋体][size=14.0000pt]溶剂[/size][/font][font=宋体][size=14.0000pt][font=宋体]问题之[/font] [/size][/font][font=宋体][size=14.0000pt]-[/size][/font][font=宋体][size=14.0000pt] 溶剂与[/size][/font][font=宋体][size=14.0000pt]固定相的相容[/size][/font][/align][font=宋体][size=12.0000pt][font=宋体]概述:[/font] [font=宋体]采用不分流进样操作方法时,溶剂聚焦是常用的改善色谱峰柱效的手段[/font][/size][/font][font=宋体][size=12.0000pt]。在此情况下,一定[/size][/font][font=宋体][size=12.0000pt]要考虑溶剂和色谱柱固定相的相容问题。[/size][/font][align=center][font=宋体][size=12.0000pt][font=宋体]一[/font] [font=宋体]常见的不分流进样条件下的柱温程序[/font][/size][/font][/align][font=宋体][size=12.0000pt][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]在使用不分流进样条件时,参考文献[/size][/font][font=宋体][size=12.0000pt]中[/size][/font][font=宋体][size=12.0000pt]往往会给出类似如[/size][/font][font=宋体][size=12.0000pt][font=宋体]图[/font]1所示[/size][/font][font=宋体][size=12.0000pt]的柱温程序[/size][/font][font=宋体][size=12.0000pt]:[/size][/font][align=center][font=Calibri][size=10.5000pt][img=,656,436]https://ng1.17img.cn/bbsfiles/images/2020/07/202007212340578065_863_1604036_3.png!w656x436.jpg[/img] [/size][/font][/align][align=center][font=宋体][size=10.5000pt][font=宋体]图[/font]1 [font=宋体]典型程序升温曲线[/font][/size][/font][/align][align=center][font=宋体][size=10.5000pt][font=宋体]仪器:[/font]Shimadzu [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS-TQ8050 色谱柱 Rtx-5ms 30m*0.25mm*0.25mm[/size][/font][/align][font=宋体][size=12.0000pt][font=宋体]我们以农残分析为例,使用如果按照这个柱温程序来进样的话,保留较弱[/font]DDV、甲拌磷等组分的保留时间可能会在6min以上。分析方法中较低的初始柱温似乎造成了组分保留时间的延长。[/size][/font][font=宋体][size=12.0000pt]那么分析文献[/size][/font][font=宋体][size=12.0000pt]为什么要[/size][/font][font=宋体][size=12.0000pt]这样来设定呢,是否可以提高柱箱初始温度来缩短分析时间呢[/size][/font][font=宋体][size=12.0000pt]?[/size][/font][align=center][font=宋体][size=12.0000pt][font=宋体]二[/font] [font=宋体]溶剂聚焦的原理[/font][/size][/font][/align][font=宋体][size=12.0000pt]采用较高的柱箱初始温度,往往会带来不良的分析结果,例如增大色谱峰宽、降低峰高或者色谱峰分叉。[/size][/font][font=宋体][size=12.0000pt]主要原因是不分流进样方式下,样品在衬管中运行速度[/size][/font][font=宋体][size=12.0000pt]速度低,气化时间长,造成物质的起始谱带较宽。[/size][/font][font=宋体][size=12.0000pt]因此一般[/size][/font][font=宋体][size=12.0000pt]需要采用辅助聚焦的[/size][/font][font=宋体][size=12.0000pt]方法[/size][/font][font=宋体][size=12.0000pt],溶剂聚焦就是常用的手段。[/size][/font][font=宋体][size=12.0000pt][font=宋体]溶剂聚焦实现的方法很简单,降低色谱柱的初始温度,一般会低于样品溶剂沸点[/font]10度以下。[/size][/font][font=宋体][size=12.0000pt]样品[/size][/font][font=宋体][size=12.0000pt]在进样后缓慢气化[/size][/font][font=宋体][size=12.0000pt]进入色谱柱,由于色谱柱温度较低,样品就会发生冷凝,在色谱柱固定相表面就会有一定长度的分布[/size][/font][font=宋体][size=12.0000pt][font=宋体](区域[/font]A)。在载气的推动下,沸点较低的溶剂会慢慢气化和向前运行,沸点较高的目标组分仍然溶解在溶剂中。[/size][/font][font=宋体][size=12.0000pt][font=宋体]一段时间之后,液态样品的空间分布就会变得比较窄(区域[/font]B),此时柱温迅速升高,样品气化就会得到较窄的色谱峰,这个过程即为溶剂聚焦。[/size][/font][align=center][font=宋体][size=12.0000pt][img=,690,141]https://ng1.17img.cn/bbsfiles/images/2020/07/202007212341156196_6734_1604036_3.png!w690x141.jpg[/img] [/size][/font][/align][align=center][font=宋体][size=10.5000pt][font=宋体]图[/font]2 溶剂聚焦图解[/size][/font][/align][align=center][font=宋体][size=12.0000pt][font=宋体]三[/font] [font=宋体]溶剂和固定相的相容[/font][/size][/font][/align][font=宋体][size=12.0000pt][font=宋体]虽然溶剂聚焦的实现似乎比较容易,但是在实际实验中,需要注意溶剂选择的问题[/font]——应当使用与色谱柱固定相相容的溶剂。[/size][/font][font=宋体][size=12.0000pt][font=宋体]某用户使用图[/font]1所示的分析条件进样666和ddt农残(甲醇作为溶剂),获得的谱图中666色谱峰发生了分叉,如图3所示。更换样品溶剂(正己烷)之后,故障解除。[/size][/font][align=center][font=宋体][size=12.0000pt][img=,690,292]https://ng1.17img.cn/bbsfiles/images/2020/07/202007212341355875_9748_1604036_3.png!w690x292.jpg[/img] [/size][/font][/align][align=center][font=宋体][size=10.5000pt][font=宋体]图[/font]3 不良色谱图[/size][/font][/align][font=宋体][size=12.0000pt][font=宋体]该不良色谱图的原因就是溶剂和色谱柱的不相容。如图[/font]4所示,如果溶剂不能浸润固定相(样品难以在色谱柱内部形成较为均匀的液膜),样品在色谱柱内就会发生分段的分布(区域A),相同条件下区域B的分布也会变差,最终使得色谱峰形状变差。[/size][/font][align=center][font=Calibri][size=10.5000pt][img=,690,144]https://ng1.17img.cn/bbsfiles/images/2020/07/202007212341531066_6315_1604036_3.png!w690x144.jpg[/img] [/size][/font][/align][align=center][font=宋体][size=10.5000pt][font=宋体]图[/font]4 [font=宋体]溶剂不良时色谱柱内样品的分布状态[/font][/size][/font][/align][font=宋体][size=12.0000pt][font=宋体]如果不更换溶剂,也可以采用固定相聚焦的方法来改善色谱峰形状,可以将柱温程序的低温段时间拉长或长将第一阶升温速率降低[/font]——这本质上是利用了色谱柱固定相的聚焦。[/size][/font]

[b][size=18px] 食品标准的宽泛性让执行者在执行的时候往往挠头,百思不得其解。[/size][size=18px] 比如污染物测定,按照《食品安全国家标准 食品中污染物限量》GB 2762中规定污染物限量是可食用部分中允许的最大含量水平。[/size][/b][align=left][b][size=18px][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310171123404002_4723_1645752_3.png[/img][/size][/b][/align][b][size=18px] 这也意味着在检测食品的污染物时,应该取其可食用部分来进行检测,这对于一般的食品比如带核的蜜饯类(话梅)、带骨头的肉制品类(比如鸡腿),一般都能明确地判定哪些是可食用部分。啥,你说你吃肉不吐骨头——你那是个例、特例。[/size][size=18px] 但对于一些食品来说,我们经常会挠头。中国饮食文化太过丰富,尤其两广一带号称“地上四条腿的除了板凳外都吃”,对于可食用部分的界定,真是缺乏一个权威的参考依据啊。[/size][size=18px] 比如生姜,这一般是带皮吃还是不带皮吃啊?这皮到底属于可食用部分还是不属于呢?别笑,要知道这取决了检测生姜的污染物(比如镉、铅)时要不要去皮呢。在征询了若干名同事后,我们决定:生姜不去皮。[/size][/b][align=left][b][size=18px][font='宋体'] [/font][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310171123406203_3304_1645752_3.png[/img][font='宋体'] [/font][/size][/b][/align][b][size=18px] 还有一些贝壳、螺,内脏属于可食用部分还是不属于呢?对于个体较大的螺、贝,很多人会比较讲究地去掉排泄器官部分;那对于个体小的呢——蚝仔、海瓜子,不都是整个的吃嘛。好在《食品安全国家标准 食品中污染物限量》GB 2762里对于铅、镉的限量特意做了规定,一些贝、螺要去除内脏。下图是镉的要求。[/size][/b][align=left][b][size=18px][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310171123406817_996_1645752_3.png[/img][/size][/b][/align][align=left][b][size=18px][/size][/b][/align][b][size=18px] 还有一些蔬菜的可食用部分,也难以界定。比如葱,葱的根须属于可食用部分吗?这真是有的人吃有的人不吃啊。在征询了若干名同事后,我们决定:葱带根须。也就是把葱的根须视为可食用部分。[/size][/b][align=left][b][size=18px][font='宋体'] [/font][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310171123408964_7466_1645752_3.png[/img][/size][/b][/align][align=left][b][size=18px] YES OR NO?[/size][/b][/align][align=left][b][size=18px] 能吃,还是不能吃?没想到这个问题不是哲学问题、也不是食欲问题,居然是个科学问题了。[/size][/b][/align][b][size=18px] [/size][/b]
[b]关于公开征求《生态环境损害鉴定评估技术指南 生态系统 第1部分:农田生态系统(征求意见稿)》等3项国家生态环境标准意见的通知[/b]各有关单位: 为贯彻落实《生态环境损害赔偿制度改革方案》《生态环境损害赔偿管理规定》的要求,加快健全生态环境损害鉴定评估技术标准体系,规范生态环境损害鉴定评估工作,我部组织编制了《生态环境损害鉴定评估技术指南 生态系统 第1部分:农田生态系统(征求意见稿)》《生态环境损害鉴定评估技术指南 总纲和关键环节 第3部分:恢复效果评估(征求意见稿)》《生态环境损害鉴定评估技术指南 总纲和关键环节 第4部分:土壤生态环境基线调查与确定(征求意见稿)》等3项国家生态环境标准,现公开征求意见。征求意见稿及编制说明可登录我部网站(http://www.mee.gov.cn/)“意见征集”栏目检索查阅。 各机关团体、企事业单位和个人均可提出意见和建议,有关意见请书面反馈我部(电子文档请同时发至联系人邮箱)。征求意见截止时间为2022年10月8日。 联系人:生态环境部法规与标准司杨阳 电话:(010)65645281 传真:(010)65645260 邮箱:sunhaipeichang@mee.gov.cn 通信地址:北京市东城区东长安街12号 邮编:100006 附件:[url=https://www.mee.gov.cn/xxgk2018/xxgk/xxgk06/202209/W020220907344731587159.pdf]1.征求意见单位名单[/url] [url=https://www.mee.gov.cn/xxgk2018/xxgk/xxgk06/202209/W020220907344731765840.pdf]2.生态环境损害鉴定评估技术指南 生态系统 第1部分:农田生态系统(征求意见稿)[/url] [url=https://www.mee.gov.cn/xxgk2018/xxgk/xxgk06/202209/W020220907344732766855.pdf]3.《生态环境损害鉴定评估技术指南 生态系统 第1部分:农田生态系统(征求意见稿)》编制说明[/url] [url=https://www.mee.gov.cn/xxgk2018/xxgk/xxgk06/202209/W020220907344733390427.pdf]4.生态环境损害鉴定评估技术指南 总纲和关键环节 第3部分:恢复效果评估(征求意见稿)[/url] [url=https://www.mee.gov.cn/xxgk2018/xxgk/xxgk06/202209/W020220907344737234025.pdf]5.《生态环境损害鉴定评估技术指南 总纲和关键环节 第3部分:恢复效果评估(征求意见稿)》编制说明[/url] [url=https://www.mee.gov.cn/xxgk2018/xxgk/xxgk06/202209/W020220907344738104436.pdf]6.生态环境损害鉴定评估技术指南 总纲和关键环节 第4部分:土壤生态环境基线调查与确定(征求意见稿)[/url] [url=https://www.mee.gov.cn/xxgk2018/xxgk/xxgk06/202209/W020220907344738862139.pdf]7.《生态环境损害鉴定评估技术指南 总纲和关键环节 第4部分:土壤生态环境基线调查与确定(征求意见稿)》编制说明[/url][align=right] 生态环境部办公厅[/align][align=right] 2022年8月30日[/align] (此件社会公开)
(了解气相色谱的核心部件 — 气相色谱柱,第 1 部分http://bbs.instrument.com.cn/shtml/20130930/4993060/)了解气相色谱的核心部件—气相色谱柱,第 2 部分上次,在本概述的第 1 部分中我们重点介绍了气相色谱柱的开发、制造和测试。在第 2 部分中,我们将讨论固定相涂层和质量控制。http://ng1.17img.cn/bbsfiles/images/2013/12/201312082133_481224_1615838_3.gif图 1. 气相色谱柱固定相的使用百分比 1][size=24px]固定相涂层的重要性[/size]固定相决定了色谱的分离度和重现性,因此涂敷步骤对于色谱柱的总柱效及其具体应用具有重要意义。固定相应当具有一致的厚度、均匀性和对内壁的粘着力。这样才能确保色谱柱在程序升温循环或溶剂和样品进样时不会发生干扰。现代的固定相通常通过交联实现固定,交联固定相比非交联固定相具有更高的稳定性,并且可以在色谱柱使用后用溶剂清洗受污染的色谱柱。根据应用选择合适类型的固定相。常见的固定相如图 1所示。最常用的固定相为聚合物固定相。为了确保当前气相色谱柱涂层所用的聚合物与数月前乃至数年前所用的聚合物相一致,许多制造商会在聚合物用于色谱柱涂层之前对其进行合成、纯化和预测试。对于共聚物而言,控制替代物百分比对于确保色谱柱具有可重现的保留特性至关重要。除去较低分子量的部分尤其重要,因为这些部分会引起色谱柱流失(尤其在高温下)。一旦认为聚合物可以接受,则应当将其与适当的溶剂混合至合适的浓度,因为这一组成将会决定固定相的膜厚度。[size=24px]成功完成涂敷的各种方法和步骤[/size]涂敷气相色谱毛细管柱的方法包括动态方法和静态方法。表 1 显示了这两种工艺如何沉积涂层,影响各个方法中膜厚度的因素以及各自的用途和优点。[table=690][tr][td][size=24px]涂敷类型[/size][/td][td][size=24px]方法[/size][/td][td][size=24px]膜沉积[/size][/td][td][size=24px]膜厚度[/size][/td][td][size=24px]优点[/size][/td][td][size=24px]用途[/size][/td][/tr][tr][td]动态[/td][td]将包含固定相的溶剂注入色谱柱的一端并在一定压力下使溶液以恒定流速穿过色谱柱。[/td][td]当溶液穿过色谱柱时,涂层会留在色谱柱上。[/td][td]受到溶液组成、表面、固定相和溶剂的物理性质及其他因素的影响。[/td][td]制造速度快。[/td][td]主要用于 PLOT 色谱柱,几乎不用于 WCOT 毛细柱。[/td][/tr][tr][td]静态[/td][td]使用溶解于一定浓度(足以得到所需膜厚度)挥发性溶剂中的固定相将色谱柱填满。然后密封一端,对毛细管进行抽吸。[/td][td]随着溶剂从毛细管柱后部被抽出,涂层将沉积到溶剂前方。[/td][td]与溶剂中固定相浓度和管路直径呈正比。[/td][td]柱重现性高。[/td][td]WCOT 毛细柱。[/td][/tr][/table]表 1. 固定相涂敷方法固定相可使用动态或静态涂敷方法,在涂敷过程中进行[i]原位[/i]固定或交联。这一工艺必须在无氧条件下进行,以确保得到稳定、惰性的固定相。在另一个方法中,可通过首先沉积单体,再通过加热或催化在管壁表面聚合,形成固定相。该工艺将固定相锁定在内壁上,然后使其固定。它还可以同动态或静态应用方法相结合。论采用何种工艺,不论色谱柱内径 (id) 多大,控制相比率 (β) 都至关重要。如果不同色谱柱的内径不同,可通过调节膜厚度以获得恒定的相比率 β。毛细管的一般容差为 +/- 6 µm,对 0.53 mm 毛细柱而言,差异为 1.1%;但对 0.25 mm 毛细柱而言,差异则为 2.4%。待固定相完成键合或固定后,必须通过冲洗除去未反应的聚合物,然后除去清洗溶剂。可采用缓慢加热色谱柱同时通入惰性气体的方法除去溶剂。最后,在无氧气体流过色谱柱的同时将色谱柱加热至其上限温度。

固相萃取技术及应用 ——迪马科技论坛在线技术讲座 在我国,食品安全日益成为亿万消费者关注的热点。固相萃取技术是食品安全检测过程中最重要的前处理技术,为了给食品安全检测领域的各位友人以支持,迪马科技特举办本次在线技术讲座,欢迎大家积极参与并讨论。一、讲座举办时间:在线时间:2010年4月14日 14:00~16:30活动时间:2009年4月14日——2009年4月20日奖品发放时间:2009年4月26日二、讲座举办地点:仪器信息网——论坛——行业应用——食品监测(食品检验检疫)三、讲座内容:1 主讲人:godblesschina(迪马技术工程师)2 主题:固相萃取技术及应用 内容提纲第一部分 引言第二部分 固相萃取过程中涉及的作用力第三部分 ProElut SPE产品介绍第四部分 ProElut SPE产品的优势第五部分 固相萃取方法的建立第六部分 SPE常见问题及解决方法第七部分 固相萃取技术分析案例
成长的很大一部分,是接受。接受分道扬镳,接受世事无常,接受孤独挫折,接受突如其来的无力感,然后发自内心地去改变。
请问大家,“绝大部分”,“大部分”,“部分”,“小部分”如果用百分比来表示应该是多少?或者范围是多少?谢谢!
氯化钴是大家非常熟悉的一种物质,实验室用到的硅胶干燥剂里一般有添加氯化钴用作吸湿指示剂,硅胶干燥剂吸水变红烘干变透明便是氯化钴的作用。最近有部分企业提出要限制电子产品中限用氯化钴。那么氯化钴的危害是什么?有哪些法规在管控?氯化钴除了应用于硅胶干燥剂以外,还会应用于哪些材料?[color=#DC143C][size=4][B]另:欧盟在2008年6月18日颁布第一版REACH高关注物质候选物质列表中有列入氯化钴[/B][/size][/color]
( 本文只是一种探讨交流,可能有不足不妥之处,欢迎批评指正。未经同意,请勿转载。多谢合作!)保留指数应用(七)保留指数估算(1)----非极性或弱极性柱子上面部分化合物的保留指数的估算保留指数作为气质定性一个强有力的辅助手段,在天然香精油,香气香味材料,香精产品等的分析鉴定中广泛应用。当然应用范围远不止这些。对于异构体,同系物和结构特征相似的化合物,由于其质谱图非常相似,谱库检索结果匹配度,排列次序都很接近,检索给出的顺序也不一定正确。但它们的保留时间可能会不同,但保留时间只能在特定色谱条件下不变,而保留指数在固定相相同下有可比性。虽然在相同的柱子上和相同的色谱条件下,两个不同的化合物的保留指数有可能相同。但两个化合物同时具有相同的保留指数(或保留时间)和相同的质谱图的不可能性极小。虽然保留时间也可以帮助确认,但保留时间会随着柱子使用的不同阶段新旧等因素而变化,但保留指数是和固定相为主要因素的一个值,相对比较固定不变。所以才有保留指数辅助定性更具有优势。在谱库检索的基础上,用保留指数来确认结果。是一种很重要的手段。 一、基本概念保留指数retention index或Kovats Index(RI或KI)概念是由Kovats在1958年提出。是把组分的保留值用两个分别前后靠近它的正构烷烃来标定(这比仅用一个参比物质的相对保留值定向更为精确)。正构烷烃的保留指数规定为等于该烷烃分子中碳原子数的100倍。例如正己烷的RI为600,正庚烷为700,正十五烷为1500.正构烷烃的RI与所用的色谱柱,柱温及其它操作条件无关。 保留指数(RI)的计算公式如下:I=100Z+100R(x)- logt’R(z)]/R(z+1)- logt’R(z)] (恒温分析)式中:t’R为校正保留时间;Z和Z+1分别为目标化合物(X)流出前后的正构烷烃所含碳原子的数目;这里:t’R(z) t’R(x)







 我要推广仪器
我要推广仪器
 下载APP
下载APP