今天测砷标曲做不出来,前两天做出汞的标曲,今天机子稳定性还不错,空白三四次就稳定了,但是标曲不论是人工稀释还是手动稀释都不行,还原剂是按照国标的方法配置,载流是2%的硝酸,是哪里出了问题?
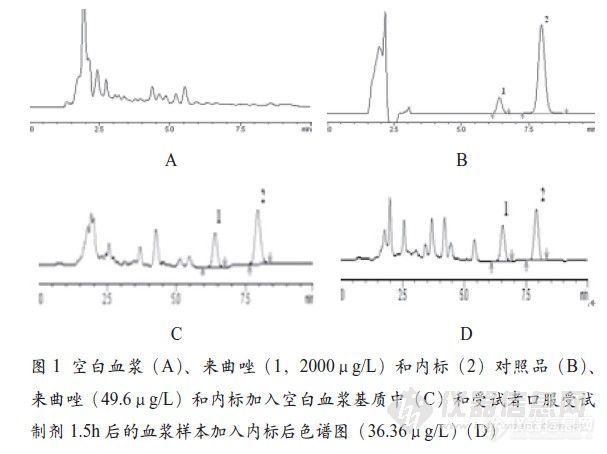
【作者】 赵玉婷; 徐世希; 王胜峰; 周彦彬; 田娟; 冉黎灵; 左英; 丁劲松;【Author】 ZHAO Yu-ting, XU Shi-xi, WANG Sheng-feng, ZHOU Yan-bin, TIAN Juan, RAN Li-ling, ZUO Ying, DING Jin-song (School of Pharmaceutical Sciences, Central South University ,ChangSha 410013, China)【机构】 中南大学药学院;【摘要】 目的建立测定人血浆中来曲唑浓度的高效液相色谱法,并研究来曲唑分散片在正常人体中的生物利用度。方法血浆样品经乙醚萃取浓缩后,用DiamonsilC18柱(4.6mm×200mm,5μm)分离,咪唑斯汀为内标,在239nm处波长处检测。采用两周期随机交叉给药方案,18名受试者分别单剂量口服2.5mg来曲唑分散片(受试制剂,T)或来曲唑片(参比制剂,R),不同时间点取血、测定血浆样品中来曲唑浓度,比较二者生物利用度。结果来曲唑和内标色谱峰完全分离,血浆内源性杂质不干扰测定。来曲唑浓度在2.48~99.2μg/L范围内与峰面积比线性关系良好,其定量下限为2.48μg/L。方法回收率在87.88%~104.02%(n=20),批内和批间相对标准偏差(RSD)均0.05)。... 更多还原【Abstract】 Objective To develop and validate a high-performance liquid chromatography (HPLC) method for the analysis of letrozole in human plasma and to study the relative bioavalability of letrozole dispersible tablets versus tablets. Method The liquid-liquid extraction of letrozole from plasma was performed with aether. the letrozole was chromatographed on a Diamonsil C18 column(4.6mm×200 mm, 5μm) with a mobile phase consisting of acetonitrile and phosphate buffer in the ratio of 41∶59v/v, the mobile pha... 更多还原【关键词】 来曲唑; 高效液相色谱法; 相对生物利用度; 【Key words】 Letrozole; High-performance liquid chromatography; Relative bioactivity; http://ng1.17img.cn/bbsfiles/images/2012/08/201208201100_384557_2352694_3.jpg

现在在做水中Fe元素,但标曲一直走不出来,给工程师联系,排除了各种可能,给工程师看了谱线搜索,说波长不准确,需要校正。后来又把所有电源线拔了重新开机,自检,走了一遍还是不行,就是反复出现如下图的情况。做Mn元素波长也有偏差,但标曲走的还行,如下图。就是Fe元素一直走不出来标曲,以为是标液问题,又重新配了一遍还是不行,母液是新拆开的,试着走了一下母液,吸收值还是负数,不知道到底是哪儿的问题。求大神指教:1.自己是否可以校正波长?该如何校正?2.做Fe元素有没有方法可以探讨?3.有没有大神可以解释一下这种情况该如何结果?最后我昨晚做梦都是在做Fe 马上要崩溃了 求大神指导![img=,690,516]http://ng1.17img.cn/bbsfiles/images/2018/03/201803141100517406_8080_3350919_3.png[/img][img=,690,1226]http://ng1.17img.cn/bbsfiles/images/2018/03/201803141100521876_7960_3350919_3.png[/img][img=,690,516]http://ng1.17img.cn/bbsfiles/images/2018/03/201803141101177956_1420_3350919_3.png[/img][img=,690,516]http://ng1.17img.cn/bbsfiles/images/2018/03/201803141105370946_9404_3350919_3.png[/img][img=,690,388]http://ng1.17img.cn/bbsfiles/images/2018/03/201803141106284793_7107_3350919_3.png[/img]
求原子荧光做汞砷硒锑铋技巧,标曲可以提前配好放冰箱吗?每次拿出来跑一下标曲。
做山嵛酸甘油酯的镍,灰化1100,原子化2700,做的标曲总是不好,10ppb的主浓度,信号值分别为0.076 0.104 0.132 0.158 0.182 空白为0.033,r只有0.98,大家都是怎么做的啊,给个建议我
请问用固相微萃取或顶空法来做原油的分析可行性如何?
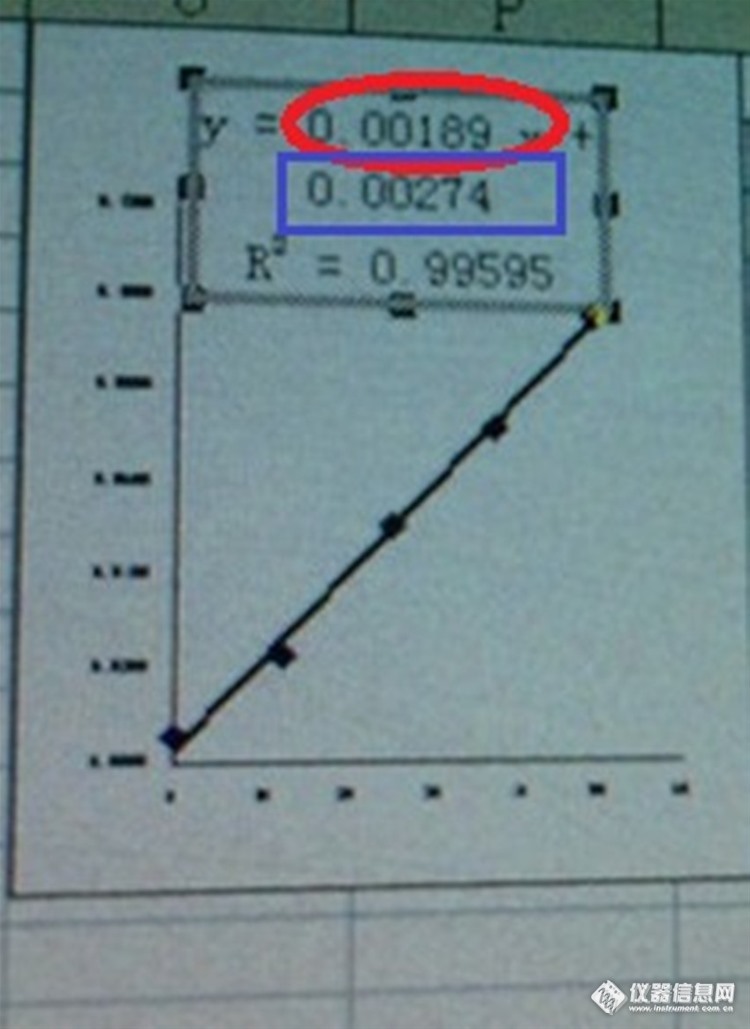
如题。最近发现仪器做的标曲跟自己在EXCEL上做出来的标曲有所差别,不知是怎么回事?难道用最小二乘法做出来的标曲还会不一样?看来两者不是用同样的方法。哪位老师为我们解释一下好吗? 在下图中,仪器做出来的标曲方程:A=0.00197-0.00044,而EXCEL做的标曲方程:y=0.00189x+0.00274,斜率和截距都有差别。呃,这两者是同一组数据做出来的这一点是肯定的http://simg.instrument.com.cn/bbs/images/default/em09511.gifhttp://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_669869_2076515_3.jpg图1 仪器系统做的标曲 斜率:0.00197 截距:-0.00044http://ng1.17img.cn/bbsfiles/images/2016/12/201612132024_01_2076515_3.jpg图2 EXCEL做的标曲 斜率:0.00189 截距:0.00274
气质联用检测空气中VOCs热解析进样标曲做不出来是怎么回事,请教各位前辈。我这边是使用国产热解析进样,但是标准曲线做不出来,有没有做过相关项目的前辈给我指导下,谢谢
,我这边用GCMS做标曲,跑出来的峰谱图连它证书上的物质都没能找的到可能是什么原因,有什么方法可以解决?做邻苯6p的标曲只找到BBP DBP DEHP其他没找到啊!标准物质是振翔买的500ppm的,浓度是50ppm 最高
我们学院18年12月到了一台[url=https://insevent.instrument.com.cn/t/yp][color=#3333ff]ICP-MS[/color][/url](PerkinElmer NexlON 2000),我们课题组测重金属元素刚好要用,由于学院是第一次有这个仪器,对于出现的好多问题求助无门。特来询问各位大神,[url=https://insevent.instrument.com.cn/t/yp][color=#3333ff]ICP-MS[/color][/url]数据应该怎么看?如何处理?测量中出现的负值应该怎么办?还有就是用什么值来做标曲?往不吝赐教,在下感激不尽。
做SPME时候,样品的泡沫较多,可能会碰到萃取头。请问有什么好办法来解决?
土壤氟化物的标曲,完全按照HJ837-2017为什么做不出来,柠檬酸钠是沪市分析纯,前三个点5.18.20微克,浓度50mg/L,5的测得电位值283
群友提问: 想问一下大家,做有机磷农药,一般做标曲的时候用哪几个浓度的点比较好呢?0.03 0.05 0.1 0.2 0.3。这几个点好不好,做有机氯农药检测,同样用这几个浓度点做标曲,可否?如何参考检出限,来确定标曲所用的浓度点呢?
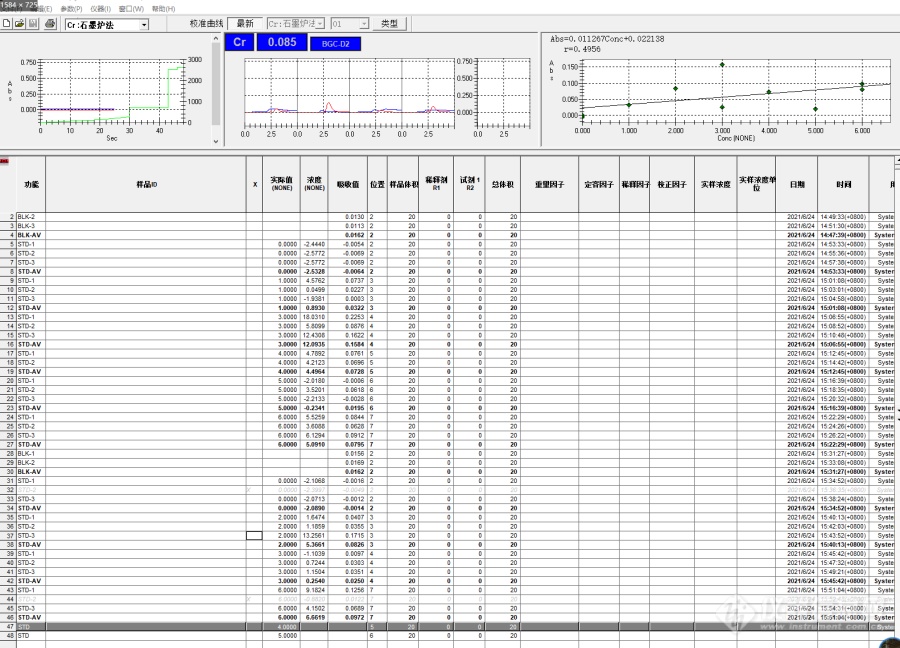
[img=,690,463]https://ng1.17img.cn/bbsfiles/images/2021/06/202106241612304744_1119_3054767_3.png!w690x463.jpg[/img]我用的是岛津AA—7000[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原吸[/color][/url] 这个仪器从来 的时候做Cr 不是空烧没值,就是做标曲 没值, 标曲浓度一般为(1,2,3,4,5)ng/mL, 做下来 就是五个点浓度毫无规律,乱七八糟。后来修改了升温程序,好了一段时间,现在又出问题了。标曲做不出来,就是一个标曲点测三次数值也是乱跳。一直找不到原因,有没有哪位老师能帮帮我?石墨炉原点位置也调整尝试过了。[img=,690,534]https://ng1.17img.cn/bbsfiles/images/2021/06/202106241603194080_2215_3054767_3.png!w690x534.jpg[/img][img=,690,496]https://ng1.17img.cn/bbsfiles/images/2021/06/202106241603290763_3367_3054767_3.png!w690x496.jpg[/img]
打算以峰面积和浓度来做标准曲线来定量,想请教一下应该做直线标曲呢还是曲线标曲呢,还有要取点时是2倍、4倍、6倍、8倍、10倍取呢,还是1、2、4、8、16这样取呢,谢谢!
我这个样品是稳定的,但是仪器出问题了,导致原来的峰测不到了。之前做的方法确证稳定性什么的都有,但当时是第一次做,忘了得随行配一条标曲?°(°ˉ??ˉ?°)°? 现在可以用之前的标曲来定量稳定性的数据吗?算不算数据造假呢
[color=#333333]按着国标来,试剂空白和标曲跑出来的峰都是一样的,没有表明是甲醇和叔戊醇的峰是怎么回事啊,求问啊[/color]
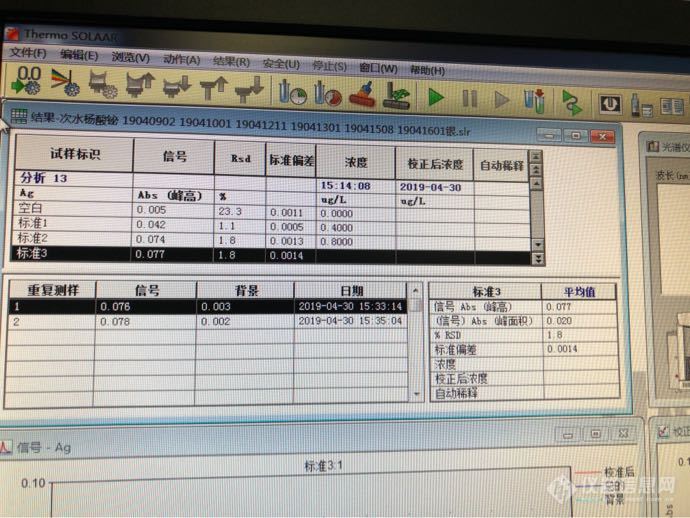
请教大家,我这个是怎么回事呀,一直第三个标准做不上去,换了好多温度,到了第三个标准吸光度就跟第二个一样了,什么原因啊,换了新的石墨管,洗了进样针,准直了进样针,找不出来原因了[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/04/201904301532564506_7746_2567134_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/04/201904301533124503_2074_2567134_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/04/201904301533347927_9916_2567134_3.png[/img]
做纺织品甲醛,取5ml样品萃取液,加5ml纳氏试剂,测出来的溶液浓度是变成萃取液的一半吗
各位站友,我测一个化学药在动物组织中的分布,如果用匀浆上清液做标曲,应该就不要测基质效应了吧?我看很多人是用匀浆上清来做的。谢谢~
各位站友,我测一个化学药在动物组织中的分布,如果用匀浆上清液做标曲,应该就不要测基质效应了吧?我看很多人是用匀浆上清来做的。谢谢~
用的是Thermo 的GC色谱仪按照国标GBT 5750.8 方法做,从同一瓶三氯甲烷四氯化碳混标出来的标准系列5个点(0.2—5 ppb),线性为,0.91;达不到0.99孵化池40℃平衡了1小时,进样量0.3mL,色谱条件和标准一样,出来的峰型很好顶空瓶120℃加热了2h,密封垫圈也用煮沸的水洗过晾干衬管换下来清洗过,柱子应该是没问题的milli-Q超纯水机出来的水检出很高的三氯甲烷四氯化碳响应,故换用了市售屈臣氏蒸馏水(低检出),煮沸的水四氯化碳响应反而变高怎么才能把这个标曲做出来?已经弄了4次了,最好一次是0.91,最坏一次完全没线性。而且空白略高
rt 和积分报告里面的峰面积不一样啊 如果我自己计算标曲 应该用哪个值作为纵坐标呢?
如何做一维H的去偶谱,来判断较复杂的氢之间的耦合关系?
我做总磷和正磷的水化,可是我的正磷总是检测不到,在0.00以后的位置,而国标检测的下限是0.01mg/l,这么说 就都检测不到了,而且我做正磷标曲的浓度最低也有0.01吧,这么说我的标曲就没有意义了,如果想检测出来,有什么其他的办法么?急,多谢
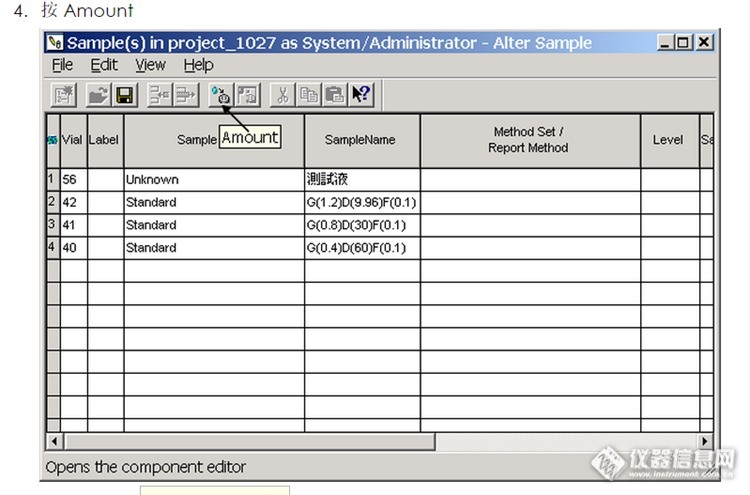
Hello,亲们!我是waters液相的新手一枚,虽然之前有岛津系列液相的小小经验,但是因为工作需要开始使用waters的液相,结果就是最近2个月有点疯掉了。。。。。。http://simg.instrument.com.cn/bbs/images/default/em09509.gif 首先,因为是二手仪器,有一些小配件,管路之类的要自己安装。工程师安装好了99%,但是就是那不到的1%,就快泪崩。缺少指导书,缺少经验在情况下只能在液相色谱之家求助大神,外加自己摸索。终于仪器开始使用了。。。。。http://simg.instrument.com.cn/bbs/images/default/em09505.gif 因为公司要求,我们使用的是Empower的英文软件,经过各种摸索和度娘的帮助,磕磕绊绊中仪器运转起来(其中有不少辛酸泪)。而且刚开始几乎每天都能遇到不少小问题,比如电脑连不上机,开机压力过高,分析样品峰形不好甚至怪异(原谅我太水了,已在提高中)等等。每天接踵而至的问题让我有点无奈又急躁,我只有自己在论坛和液相色谱之家一遍遍呼唤,然后问题大都能得到很好的解决。。。。。http://simg.instrument.com.cn/bbs/images/default/em09510.gif 前几天又遇到一个问题了——就是制作标曲。最开始就是各种找资料,一步一步按照资料走,失败,重新尝试一次,还是失败。于是我想到的液相色谱之家的那群可爱的大神。。。。。药厂小QC-kanhama给了我很大的帮助,他说: “你先设积分pro 能够自动积到你要的峰 再在alter samp里把samp type改成std 调选刚才的积分pro 把浓度填进去 保存 最后在channel里右击积分 清除校准曲线 回车 进result看看结果。”中间还教我很多东西,以及我的尝试之后,制作标曲还是失败。。。。。。最后他很无奈的说了句,你自己去找教程吧。。。。 在快下班的时候,我突然想到好像标曲是要2D谱图才行,我跑样时选择的3D的,这样当然不能做出曲线了。于是第二天重新跑了一系列梯度的2D图,重新按照教程来,仍然失败了。。。。。。http://simg.instrument.com.cn/bbs/images/default/em09504.gif(oh my god!到底是为什么?一定是上辈子做了太多错事,这辈子Waters来惩罚我!)经过很多次的不同尝试,我终于发现了关键的一个步骤,找到失败的原因,但为什么很多教程都没有提到?为了帮助一样的Waters新手,让大家少些烦恼,所以我想把我制作标曲的成功的关键步骤告诉大家。希望大神们也给我指正!基本的步骤就是大多数的教程,也就是药厂小QC-kanhama教我的那样,但是关键一步是在Alter Sample 的一个小细节。 将标准品反黑,进入Alter Sample准备填入浓度,http://ng1.17img.cn/bbsfiles/images/2017/01/201701191700_667746_3083385_3.png按Amount,http://ng1.17img.cn/bbsfiles/images/2017/10/2016041522144741_01_3083385_3.png填入浓度及Component Name后你需要在Edit界面下的 Copy Compoment From Methodset亲亲点击一下,http://ng1.17img.cn/bbsfiles/images/2016/04/201604152216_590488_3083385_3.jpg在按照一般教程走完,你就能得到你想要的标曲了。 看起来是不是很简单,可是我却摸索了好多次才成功。。。。。http://simg.instrument.com.cn/bbs/images/default/em09511.gif希望我的提醒对新手们有帮助吧,也希望大家原谅我的啰嗦(主要是抱怨和感谢)。。。。。。最后感谢仪器信息网,感谢液相色谱之家的各位大神!希望我们度过愉快的周末再去迎接以后的挑战!http://simg.instrument.com.cn/bbs/images/default/em09503.gif
用mtbe做萃取,但是没有合适的离心机来离心现有的玻璃瓶,我可以就用聚丙烯试管作萃取吗? 还是可以用玻璃瓶萃取,然后等它自然沉淀,不离心了? 谢谢了
前两天做实验,称量硼氢化钾时,把坨状的硼氢化钾用钥匙敲碎,取药称量,后来做实验发现标准曲线做的挺好(四个九),质控样做不准了,完全不在偏差范围内,后来上网查了下,知道应该是硼氢化钾潮解了,想问下各位大佬,硼氢化钾潮解了对标准曲线和测质控样,结果有什么影响(1,硼氢化钾潮解做出来的标准曲线相对于真值是偏小了还是偏大了,换句话说用这个曲线去测一个标准的10微克/升砷标液,测出来的浓度偏大还是偏小 2、硼氢化钾潮解做出来的标准曲线去测质控样结果会怎样)?
我之前用的凡士林做润滑剂,但是在做GC/MS检测的时候发现凡士林溶到有机溶剂中被检测出来了好多杂峰.为了不出现凡士林杂峰的干扰,我打算不用润滑剂了,但是在萃取时又出现了新问题,就是没有润滑剂就不密封了,溶液会一点一点漏出来.看来还是需要有点润滑剂的,但是就不知道该用哪种比较好,能够尽可能不引入杂质. 有用液液萃取作为GC/MS前处理的同志是怎么做的?
在做水质和饮用水挥发酚用4-氨基安替比林方法时,曲线做得出来,但盲样总做不出来,曲线和盲样需要有蒸馏过程吗?还有计算盲样时减去曲线点空白计算吗?还是减去纯水空白计算?







 我要推广仪器
我要推广仪器
 下载APP
下载APP