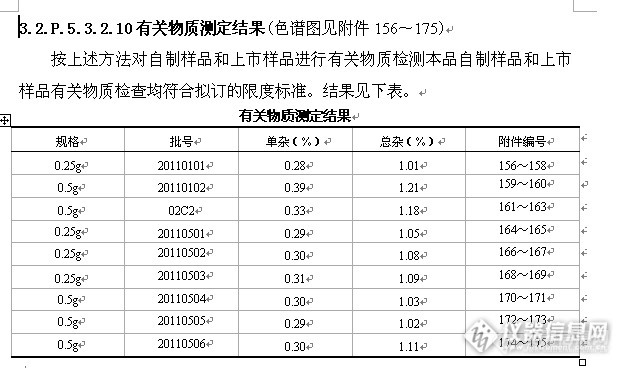
继“宝刀未老,刀走偏锋,阿莫西林舒巴坦匹酯片含量测定方法学部分”,根据该项目整理后,于是“接步献刀”进行的有关物质方法学研究,本品国内外药典均无收载且是复方制剂,根据相关的转正标准进行试验,均不理想,对有关物质的研究有一定的难度,根据相关资料进行研究,下文主要简介比较重要的部分如流动相的摸索和耐用性试验以及波长的选定,其他的同前几篇,具体如下:项目:有关物质(3.2.P.5.2.4)检查方法:HPLC法试验条件:仪器:LC-10AT VP(SHIMADZU) SPD-10A VP(SHIMADZU)万分之一电子天平(Sartorius ABS-124S型)工作站(LCsolutionlite色谱工作站)色谱柱(填料:C18,规格:250mm×4.6mm,填料粒径:5μm)Wel5184425,sn:w10212096,ln:w1801.19UV检测器(210nm)柱温:室温流动相:0.01mol/L十二烷基硫酸钠溶液(用磷酸调节pH值至2.5±0.02)-甲醇(45:55)为流动相流速:1.0ml/min运行时间:约55分钟系统适用性:理论板数按阿莫西林峰计算不低于2000。具体试验操作:取本品细粉适量,精密称定,用流动相适量溶解并稀释制成每1ml中约含阿莫西林和舒巴坦均为0.5mg的溶液,摇匀,滤过,取续滤液作为供试品溶液;精密量取供试品溶液1ml置100ml量瓶中,用流动相稀释至刻度,作为对照溶液。取对照溶液10μl注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高为满量程的20%~25%。再精密量取供试品溶液和对照溶液各10μl,分别注入液相色谱仪,记录色谱图,对照溶液中阿莫西林的峰面积As,供试品溶液中各杂质的峰面积Ai均通过自动积分法测定,以各杂质峰面积与对照溶液阿莫西林峰面积的比值计算得出各杂质的含量,总杂质为各杂质的和。计算公式:各杂质的量(%)=Ai/As杂质总量(%)=∑ihttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300029_448425_1621890_3.png3.2.P.5.3.2.1 流动相选择该品种目前在中国药典、美国药典、日抗基和英国药典均未收载。参照新药转正标准第55册收载的阿莫西林舒巴坦匹酯片质量标准WS1-(X-145)-2004Z、进口药品注册标准JX20080076、舒巴坦匹酯国家质量标准WS-516(X-439)-2001(试行)以及阿莫西林舒巴坦匹酯制剂的其他相关研究资料(傅小雅.HPLC-DAD法测定阿莫西林舒巴坦匹酯分散片中舒巴坦匹酯含量及有关物质.中国药房2008年第19卷第13期:1011-1012;姜红,胡昌勤,金少鸿.阿莫西林-舒巴坦匹酯片含量及有关物质质控分析方法的建立.药物分析杂志2002,22(2):91-94)进行有关物质检查流动相选择。由于阿莫西林和舒巴坦匹酯极性差别较大,通常会造成阿莫西林在死时间附近出峰,而舒巴坦匹酯出峰时间较迟。本品试验表明流动相加入十二烷基硫酸钠溶液能有效检查有关物质。初步拟定流动相体系及试验结果统计见下表。http://ng1.17img.cn/bbsfiles/images/2013/06/201306300030_448426_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300031_448427_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300031_448428_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300032_448429_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300033_448430_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300033_448431_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300034_448432_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300035_448433_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300035_448434_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300036_448435_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300037_448436_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300038_448437_1621890_3.png3.2.P.5.3.2.4有关物质检查波长选定(色谱图见附件115~117)本品目前在中国药典、美国药典、日抗基和英国药典均未收载。参照新药转正标准第55册收载的阿莫西林舒巴坦匹酯片质量标准WS
蜂蜜中吠喃它酮、吠喃西林、吠喃妥因和吠喃唑酮[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=83718]蜂蜜中吠喃它酮、吠喃西林、吠喃妥因和吠喃唑酮[/url]
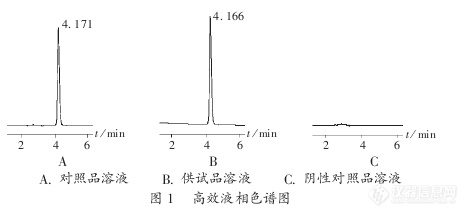
作者:http://d.g.wanfangdata.com.cn/Images/head_pic.gif邓芳 http://d.g.wanfangdata.com.cn/Images/head_pic.gif万莉 http://d.g.wanfangdata.com.cn/Images/head_pic.gif崔小平 Author:Deng Fang Wan Li Cui Xiaoping 作者单位:重庆市涪陵区中心医院,重庆,408000 重庆市万州药品检验所,重庆,404000 重庆三峡中心医院,重庆,404000摘要: 建立呋喃西林溶液中呋喃西林含量的高效液相色谱法.方法 色谱柱为Diamonsil C18柱(250 mm x4.6 mm,5μm),流动相为甲醇-水-三乙胺(40:60:0.1),检测波长为254 nm,流速为1.0 mL/min.结果 呋喃西林进样量的线性范围为O...http://ng1.17img.cn/bbsfiles/images/2012/08/201208201647_384759_2379123_3.jpg
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=102808]HPLC—DAD法测定阿莫西林舒巴坦匹酯分散片中舒巴坦匹酯含量及有关物质[/url]

因本品按照以前的转正标准,无法达到分析要求,于是刀走偏锋,另辟蹊径,以下是本品含量测定方法学研究内容:项目:含量测定(3.2.P.5.2.9)检查方法:照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定试验条件:仪器:LC-10AT VP(SHIMADZU) SPD-10A VP(SHIMADZU)万分之一电子天平(Sartorius ABS-124S型)工作站(LCsolutionlite色谱工作站)色谱柱(填料:C18,规格:250mm×4.6mm,填料粒径:5μm)Wel5184425,sn:w10212096,ln:w1801.19UV检测器(210nm)柱温:室温流动相:磷酸二氢钾溶液(取磷酸二氢钾3.06g,加水900ml溶解后,用磷酸调节pH值至3.0,再加水稀释至1000ml,混匀)-乙腈(40:60)。流速:1.0ml/min运行时间:约10分钟具体试验操作:取本品20片,精密称定,研细,精密称取细粉适量(约相当于阿莫西林和舒巴坦分别为50mg),置100ml量瓶中,加流动相适量,超声使阿莫西林和舒巴坦匹酯溶解,加流动相稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液;另分别取阿莫西林和舒巴坦匹酯对照品适量,加流动相制成每1ml中分别含0.5mg阿莫西林和舒巴坦的溶液,作为对照品溶液。精密量取供试品溶液和对照品溶液各10μl,分别注入液相色谱仪,记录色谱图,按外标法以峰面积计算,即得。计算公式:标示量百分含量(%)=××100%式中:Cs为对照品的浓度(mg/ml);At为供试液中相应主峰面积;Nt为供试液的稀释倍数;AS为对照品溶液中相应主峰面积;W为供试品取样量(mg)。http://ng1.17img.cn/bbsfiles/images/2013/06/201306292348_448407_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292348_448408_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292349_448409_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292350_448410_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292350_448411_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292351_448412_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292351_448413_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292352_448414_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292353_448415_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292353_448416_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292354_448417_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292354_448418_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292355_448419_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292356_448420_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292357_448421_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292358_448422_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292358_448423_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292359_448424_1621890_3.png
来源:中国医药报因受环保压力的影响,近来国内多家青霉素原料药生产企业包括联邦制药彭州生产基地、阿拉宾度(大同)生物药业、四川制药等都处于停产或大幅减产状态,使青霉素工业盐的市场价格大幅攀升。目前,原料药价格涨潮已进一步传导到商业渠道——本月以来,青霉素工业盐的下游制剂产品阿莫西林已在全国范围内普遍提价。 “阿莫西林的采购价已经涨了20%左右,几乎要把进销差价抹平了。”广东金康大药房总经理郑浩涛说,“如果涨价再持续一段时间,我们就要考虑终端提价了。”在全国其他一些地区,零售药房已开始提高阿莫西林终端售价。 同时,笔者从全国三大阿莫西林生产厂家中的四川蜀中制药和石药集团中诺药业获悉,目前在全国范围内都出现了阿莫西林出厂价提价现象。“有些企业的产品价格甚至提高了30%。”蜀中制药常务副总经理刘文武说。 此番提价的主要是低端普药阿莫西林胶囊和注射剂,并不包括阿莫西林/克拉维酸钾、阿莫西林/舒巴坦钠等复方制剂。 根据价格监测统计,阿莫西林原料药已从低潮时期不到200元/千克涨至目前300元/千克左右。“现在有的原料药厂的报价甚至达到了330元/千克。”中诺药业总经理李猛说,原料药成本大概占到该制剂成本的80%。 “我们正在计划暂停原料药的采购,现在的采购价太高了,我们目前的原料储备还可以用到6月份。”刘文武说。 而中诺药业和哈药集团制药总厂则不同,这两家企业由各自的集团供应原料药。 “依靠外部供应原料的企业确实比较难受,对小企业来说不只是原料价格高的问题,而是根本就没有原料可供采购。”李猛说。据悉,目前阿莫西林上游中间体6APA正处于脱销状态,下游制剂企业根本不敢轻易下订单。 据了解,阿莫西林是目前应用广泛的抗感染类药物。此前阿莫西林已过了“黄金时期”,其价格低廉,国内竞争非常激烈,生产企业多达两三百家,年产量接近200亿粒。经历多轮的国家降价和企业间的价格战,阿莫西林的利润原本已极其微薄,如0.25克×24粒包装的阿莫西林胶囊零售价已由前几年的10多元/盒降至4元/盒,50粒装零售价也基本上在5~7元左右,比国家制定的零售价还低很多。 “阿莫西林生产的集中度正在提高,目前生产量最大的前三家企业的产能每年大概在120亿粒,占有60%的市场份额。”李猛说:“下一步,几大厂家不再打价格战了,供求将会趋于稳定。”但据李猛估计,上游货源紧张的问题可能要持续到今年下半年,而目前阿莫西林原料药的需求量仍然保持上升势头。
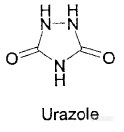
呋喃西林代谢物产生途径的研究(烟台杰科检测服务有限公司,山东 烟台 265231)摘要:呋喃西林代谢物(SEM)是检测呋喃西林的标记物,本身具有致癌和弱毒性。研究发现食品中SEM的来源途径多样,不仅限于呋喃西林原药在动物体内的代谢,还包括偶氮甲酰胺(ADC)高温热解、次氯酸钠与含氮物质反应等。关键词:呋喃西林代谢物(SEM);偶氮甲酰胺(ADC);次氯酸钠 ;标记物Nitrofurazone metabolites produced by a variety of ways 关键词:呋喃西林代谢物(SEM);偶氮甲酰胺(ADC);次氯酸钠 ;标记物呋喃西林(图一)是一种引入硝基的广谱抗菌类药物, 因其杀菌能力强、抗菌谱广、不易产生耐药性、价格低廉、疗效好等优点,得到广泛应用。呋喃西林在临床上表现为明显的三致作用(致癌、致畸、致突变),因此引起各国的高度重视,欧盟早在1995年就禁止呋喃西林用作兽药,澳大利亚、美国也相继在2001年和2002年出台了相关法律,将呋喃西林作为养殖禁用药物。 呋喃西林在动物体内极易降解,短短数小时内即可代谢为呋喃西林代谢物(SEM)(图二),SEM与蛋白质结合,性质较稳定不易分解,可在动物体内存留数周。SEM常被用作检测呋喃西林原药的标记物,世界上大多数国家都以监测SEM来达到对呋喃西林原药监控的目的。 研究者发现,食品中SEM的来源不仅限于呋喃西林在动物体内的生物代谢,偶氮甲酰胺的高温热解反应、次氯酸钠作为消毒剂在食品加工过程中与含氮物质接触等都有可能导致SEM的产生。根据近年来国内外的有关研究,现将呋喃西林代谢物产生的途径做以下概述: 1、呋喃西林原药在动物体内代谢 虽然世界上大多数国家都禁止将呋喃类药物用作兽药,但因其药效和价格上的优点(上述第一段),仍有养殖者私自使用。 呋喃西林在动物体内代谢后与细胞膜蛋白结合,可在数周内保持稳定,从而延缓药物在体内的消除速度。普通的食物加工方法(如烧烤、微波加热、烹调)难以使结合态的SEM降解,经验证在弱酸条件下可以使代谢物从蛋白质中释放出来,当人食用了含有SEM的食物后,在胃酸的作用下,SEM与蛋白质解离,被人体吸收,当富集到一定量时会产生致癌的危险。 2、偶氮甲酰胺(ADC)高温热解 偶氮甲酰胺(Azodicarbonamide)(图三),简称ADC,为黄色至橘红色结晶性粉末,具有漂白和氧化双重作用,常用作面粉改良剂,可改善面团的物理操作性质及面制品组织结构 。偶氮甲酰胺能将面粉蛋白质内氨基酸的硫氢根(-SH)氧化成二硫键(-S-S-),使蛋白质链相互连结而构成立体网状结构,改善面团的弹性、韧性及均匀性,使生产出的面制品具有较大的体积,较好的组织结构。偶氮甲酰胺也是一种生产聚氯乙烯材料的发泡剂,食品玻璃容器盖子上的密封圈就是用聚氯乙烯材料制成的。 2.1 ADC—聚氯乙烯材料的发泡剂 人们第一次将偶氮甲酰胺与SEM联系在一起,是在2003年欧盟发生的一次严重的食品安全事件后。2003年欧洲食品安全局通报了一批SEM超出限量的食品,这些食品包括:果汁、果酱、蜂蜜、泡菜和消毒蔬菜、蛋黄酱,芥末,酱汁和番茄酱以及一些瓶装婴儿食品。这些食品有一个共同的特点:都是带有密封圈的玻璃或金属罐包装。欧洲食品安全局发布的调查结果为:SEM残留可能是ADC引起的。ADC用作密封圈(聚氯乙烯材料)的发泡剂,高温发泡的同时产生SEM,食物在与密封圈接触的过程中,SEM发生了迁移。 ADC的分解产物主要有气体(34%),包括氮气、一氧化碳、二氧化碳和氨,以及一些非挥法性残留物,主要是联二脲(hydrazodicarbonamide,HDC)(34%)和脲唑(urazole )(27%)。ADC在180℃-220℃的高温下加热30分钟,即可生成SEM。ADC的分解产物HDC和脲唑经加热处理可缓慢生成SEM,而同样的热处理条件下,SEM也可以生成HDC和脲唑,尽管生成的量很少。欧盟在2003年10月9曰,发布了关于SEM有害人体健康的警告,SEM具有弱毒性和致癌性。2004年1月6日,发布了2004/1/EC指令,规定在2005年8月2日后禁止使用ADC作为发泡剂用于聚氯乙烯密封垫片生产中。 2.2 ADC—面粉改良剂 Pereira et al研究发现:向不含SEM的面粉样品中添加ADC,经一定条件处理后,检出SEM 2.2 μg/kg -5.2 μg/kg。这些研究似乎证明了偶氮甲酰胺是面粉中检出西林的“罪魁祸首”。 面粉中检出氨基脲的事例并不常见,更多的是经过加热或烘烤的面制品。Becalski等研究发现:将含有ADC的面粉在特定温度下烘烤,以及用含有ADC的面粉制作成面包(经高温烘烤),都能检测出较高浓度的SEM,而同样含有ADC的面粉,不经高温处理,几乎检测不到SEM。该研究同时还发现面包中心的检出浓度要比面包外壳的稍大,经分析可能是由于面包中心的温度稍高的原因。Becalski还研究了湿度对SEM产生的影响:加水后的面粉和面包在200℃条件下烘焙,与干燥的面粉和面包同条件处理后相比,前者SEM的检出浓度要略高。这与ADC的热解产物HDC水解生成SEM需要水的条件是相符的。 在欧盟国家ADC是不允许作为面粉改良剂来使用的。同比,美国、巴西以及中国允许ADC在小麦粉中的最大添加量为45mg/kg。Anton发现,ADC添加到小麦粉中约有0.1%转化成SEM。而45mg/kg的允许添加量,显然是不安全的。 http://ng1.17img.cn/bbsfiles/images/2011/06/201106190944_300449_2177386_3.jpg http://ng1.17img.cn/bbsfiles/images/2011/06/201106190946_300450_2177386_3.jpg 图1 图2http://n
SN/T2050-2008 方法测定6种β-内酰胺抗生素的残留量,结果阿莫西林和氨苄西林的回收非常低,约20和10%左右,后来经过排查,发现是加缓冲液(pH8.5)后两组分就会显著降低,大家有没有什么好的建议?方法大概是:5g样→乙腈水(15:2)溶液40ml提取→取20ml在37℃旋蒸至近干→加25ml磷酸盐缓冲液(pH8.5)溶解→上HLB小柱萃取→乙腈洗脱→氮吹→磷酸盐缓冲液(pH7.0)溶样,过滤→上机有哪位专家做过这俩组分的方法,指导下,急盼http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif
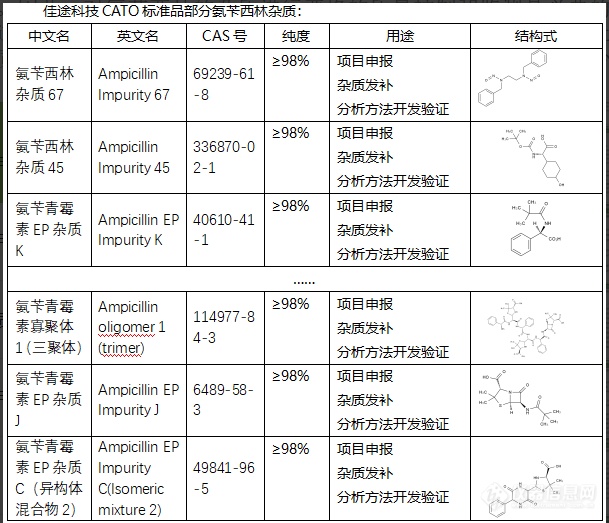
氨苄西林的杂质对于其质量、效力和安全性非常重要。一些杂质可能对药物的安全性和疗效产生负面影响。杂质的存在可能引发药物不良反应,如过敏反应,或者降低药物的效力。因此,对氨苄西林的杂质进行严格的质量控制和监测是必要的。质量控制不仅包括在生产过程中对杂质的检测和去除,还包括对贮存条件的监控,以防止在贮存过程中产生新的杂质或使现有的杂质浓度升高。CATO标准品对氨苄西林杂质的研究也有利于优化制药工艺,从而提高药物的质量,并减少不良反应和副作用的危险。这对于保证药物的治疗效果和患者安全性至关重要。[img=,609,523]https://ng1.17img.cn/bbsfiles/images/2024/02/202402052057087895_2740_6381668_3.png!w609x523.jpg[/img]
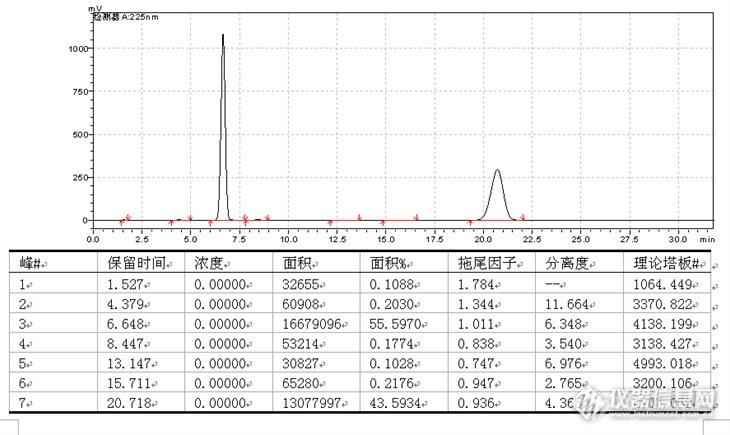
前言:这是一个老项目了,对于公开品名,是已经上报了,宝刀系列均是以前的资料,写出来和大家共享一下。由于网络问题,有些图看不到了,我在附件上显示了。建议大家看附件好了,看这个很吃力。望谅解。项目:含量测定(3.2.P.5.2.9)检查方法:照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定试验条件:仪器:LC-10AT VP(SHIMADZU) SPD-10A VP(SHIMADZU)万分之一电子天平(Sartorius ABS-124S型)工作站(LCsolutionlite色谱工作站)色谱柱(welchrom 填料:C18,规格:250mm×4.6mm,填料粒径:5μm;pn:wel518425,sn:w10212097)UV检测器(检测波长:225nm)柱温:室温流动相:流动相A为0.1mol/L磷酸二氢钾溶液-0.018mol/L十二烷基硫酸钠-甲醇-乙腈(275:275:200:250),用磷酸调节pH值至2.0;流动相B为乙腈。按下表进行线性梯度洗脱: 时间(分钟)流动相A(%)流动相B(%)090105851579010119010流速:1.2ml/min运行时间:约11分钟系统适用性:理论板数按阿莫西林峰和双氯西林峰计算应均不低于2000,双氯西林与阿莫西林的分离度应符合规定。具体试验操作:取装量差异项下的内容物,混合均匀,精密称取适量(约相当于阿莫西林25mg,双氯西林12.5mg),置100ml棕色量瓶中,加磷酸盐缓冲液(0.05mol/L磷酸二氢钾溶液-甲醇-乙腈(550:200:250)并稀释至刻度,摇匀,滤过,精密量取20μl注入液相色谱仪,记录色谱图;另取阿莫西林和双氯西林对照品,精密称定,加磷酸盐缓冲液(0.05mol/L磷酸二氢钾溶液-甲醇-乙腈(550:200:250)溶解并定量稀释制成每1ml中约含阿莫西林0.25mg和双氯西林0.125mg的溶液,同法测定,按外标法以峰面积分别计算出供试品中C16H19N3O5S和C19H17Cl2N3O5S的含量。计算公式:标示量百分含量(%)=××100%式中:Cs为对照品的浓度(mg/ml);At为供试液的主峰面积;Nt为供试液的稀释倍数;AS为对照品溶液的主峰面积;W为供试品取样量(mg)。3.2.P.5.3.6 含量测定色谱图见附件1367~1442含量测定方法学验证结果概要 项目验证结果波长选择[size=9pt
希望大咖们可以提供一款测定密闭西林瓶上方空间气体压力值的测定设备,要求测定不能破坏西林瓶状态,不会造成西林瓶中药物失效。
请教各位,阿莫西林和氨苄西林两种药物,HPLC怎么分析,包括前处理过程和液相分析方法,能否简要介绍一下大家的经验
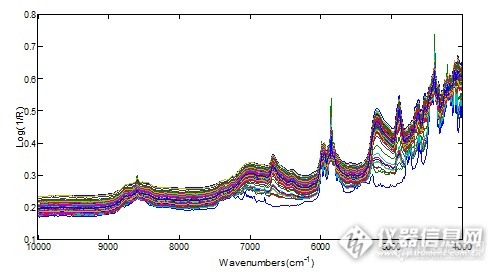
[align=center][b][url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术用于美洛西林钠舒巴坦钠药物混合过程在线混合均匀度终点监测[/b][/align][align=left][b]摘要: [/b]利用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]技术,对美洛西林钠、舒巴坦钠混合过程进行了在线监测。在研究中,分别建立了基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型,通过3个平行实验的在线混合过程,结果显示MBSD法和舒巴坦钠百分含量测定法均能有效的监测其混合过程,有效的证明了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于舒巴坦钠、美洛西林钠混合在线监测的可行性。[/align][b]关键词[/b]:[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url];分析模型;混合均匀度;在线监测自从2004年美国食品与药品监督管理局提出“过程分析技术”以来,全球的药品生产企业正在向着更高技术含量的生产方式和质量控制方式进军。近红外(Near infrared,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url])光谱分析技术因其快速,无损的特点成为“过程分析技术”的重要组成部分,是制药企业进行产品中间体质量控制的重要方法之一。传统的检测方法为高效液相色谱法,紫外可见分光光度法等需要停止混合操作时才能取样检测,并且等待检测结果所需的时间也比较长,工作效率比较低,而[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱可以进行在线检测,连续记录不同混合时间内混合物的光谱图,建立数学模型对采集数据进行分析,从而判断各组分之间是否已经达到质量均一,工作效率大幅度的提高。本研究利用 [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url] 光谱分析技术在线监测美洛西林钠舒巴坦钠的药物混合过程,从而实现混合终点的准确判断。[b]1 材料1.1试剂[/b]美洛西林钠(13102041,山东瑞阳制药有限公司)舒巴坦钠(SS201310-26,江西东风制药有限公司)[b]1.2仪器和软件[/b]AntarisII型傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url](美国ThermoFisher公司),附有积分球采样模块;RESULT采样软件;电子分析天平(Sartorius BT224S,德国);TQ数据处理软件;表面皿;药匙;自制搅拌器。[b]2 方法2.1样品的准备[/b]精密称取舒巴坦钠固体原料药10.00g,美洛西林钠固体原料药40.00g,以备进行在线混合光谱的采集。平行制备3批样品,进行混合光谱的采集。[b]2.2模型的建立[/b]目前,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于混合过程在线监测的方法可分为活性药物成分(API)定量分析模型监测和基于移动块标准偏差(MBSD)的定性分析模型监测。前者为基于API药物含量的定量监测模型,当达到混合终点时,API的含量趋于一定值,可以依据模型监测的含量是否达到理论值并趋于稳定进行混合终点的监测;后者为基于光谱的标准偏差的定性监测模型。MBSD法的基本原理为:连续采集的若干张光谱间的标准偏差变化率趋于稳定并小于限定的一阈值时可认为达到了混合终点。其具体的计算步骤为:首先确定用于计算光谱标准偏差的光谱的条数n(即移动块的宽度),当[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析仪器采集到n张光谱后计算n张光谱的峰面积(或最大峰高、平均峰高等)的标准差,当采集到n+1张光谱时将第一张光谱移除,计算最近n张光谱的标准差,如此类推,最终得到随时间变化的光谱的标准偏差,根据标准差的变化进行混合终点的监测。本研究中建立了舒巴坦钠含量的定量分析模型和基于MBSD法的定性分析模型同时对用于混合终点的判断。[b]2.3在线混合光谱的采集[/b]将称取的美洛西林钠、舒巴坦钠原料药样品放入表面皿中,然后将表面皿放在Antaris II型傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]积分球采样模块的上面,采用积分球漫反射采样方式进行光谱的采集。在运行在线混合工作流的同时采用自制的搅拌器进行样品的混合,采集得到混合过程的原始光谱,同时监测混合过程。波长范围10000-4000cm[sup]-1[/sup],每张光谱扫描次数4,混合过程中每间隔5s进行一张光谱的采集,光谱分辨率为8.0cm[sup]-1[/sup],每4个小时进行背景光谱的采集。每张[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱由1557个变量点组成。[b]2.4定量定性分析模型用于终点判断数据分析[/b]将在线混合过程进行监测,得到在线混合过程数据进行分析,以便了解混合全过程信息以及混合过程的监测。[b]2.5混合终点分析[/b]当得到混合终点时分别采集混合后的样品6处的原始[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,利用舒巴坦钠的定量分析模型预测混合终点时不同样品点处的舒巴坦钠的含量,判别是否混合均匀。[b]3 实验结果3.1分析模型的建立[/b]本研究中分别建立了在线混合过程的舒巴坦钠定量监测模型和基于移动块标准偏差的定性监测模型。[b]3.1.1 定性分析模型的建立[/b]目前混合均匀度在线监测常用的方法为MBSD法,本研究中MBSD法定性建模的参数为:选择的3个光谱区间包括全光谱、5275.6-4806.3cm[sup]-1[/sup](称为Region1)及7096.76-6344.66cm[sup]-1[/sup](称为Region2);用于计算光谱偏差的光谱的条数为5(即移动块的宽度为5)。[b]3.1.2 定量分析模型的建立[/b]本研究中所建立的定量分析模型用于监测混合过程中舒巴坦钠的百分含量的变化,因为本实验中舒巴坦钠和美洛西林钠两者间的混合比为4:1,当达到混合终点时,舒巴坦钠的百分含量应该在20%左右。其模型的具体参数见上一章中得到的舒巴坦钠百分含量的定量分析模型。[b]3.2混合在线过程数据分析[/b]本研究中平行进行了3次混合过程的在线监测,分别对3次实验结果进行分析,以充分了解混合监测过程。[b]3.2.1 第一批实验结果分析3.2.1.1 原始光谱图[/b]图1给出了混合过程中采集得到的208张原始光谱,由图中可知,处于下面的光谱较稀疏,可能属于混合刚开始的阶段,光谱会有较大的差异;处于上面的光谱较密集,其原因为随着混合的不断进行,光谱间差异越来越小,所以光谱较集中。[align=center][img=,498,274]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141912_01_1626619_3.png[/img][/align][align=center]图1 第一批混合过程原始光谱[/align][align=center] [/align][b]3.2.1.2 在线混合过程结果分析[/b]图2为定性分析模型中得到的3个光谱区间的峰面图,其中M1为全光谱建模的峰面积变化,M2为Region 1(5275.6-4806.3cm-1)的峰面积变化,M2为Region 2(7096.76-6344.66cm-1)的峰面积变化,由峰面积的变化图可知,混合过程的前100s其变化较为明显,M1不断升高,M2和M3(7096.76-6344.66cm-1)不断下降,之后峰面积值趋于稳定。[align=center][img=,525,234]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141913_01_1626619_3.png[/img][/align][align=center]图2 光谱区间峰面积图[/align]图3为舒巴坦钠含量及标准偏差变化图,由图中显示在混合的初期阶段,尤其是前100s左右,四个表征混合均匀度的参数均有着较大的变化趋势,在200-300s间四个参数有稍微较小的波动,此后随着混合过程的不断进行,表征混合均匀度的四个参数变化范围均变小,模型给出的舒巴坦钠的百分含量在20%左右,舒巴坦钠和美洛西林钠混合较为均匀,达到了混合终点。由图可知前100s是混合的主要阶段,此阶段舒巴坦钠的百分含量和标准偏差均有着明显的变化。[align=center][img=,538,292]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141914_01_1626619_3.png[/img][/align][align=center]图 3 含量和标准偏差变化图[/align][align=center](a舒巴坦钠百分含量变化 b全光谱峰面积标准差 c Region1峰面积标准差 d Region2峰面积标准差)[/align][align=left] 当达到混合终点时分别采集表面皿下6个点的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,根据建立的模型测定其舒巴坦钠的百分含量,看混合是否均匀。表2给出了用所建模型得到的6个点的舒巴坦钠的百分含量值,6个点舒巴坦钠的百分含量值在20%左右,说明混合较为均一,但是最大的值达到了22.41%,可能是由于混合装置过于简陋,加上是人为搅拌进行混合,不能达到很好的混合,部分地方没有进行很好的混合。从实验的可行性方面,初步证实了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]技术用于美洛西林钠舒巴坦钠混合的可行性。[/align][align=center]表1混合后不同点舒巴坦钠百分含量值[/align][align=center] [img=,570,70]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141915_01_1626619_3.png[/img][/align][b]3.2.2 第二批实验结果分析3.2.2.1 原始光谱图[/b]图4给出了第二批混合过程中采集得到的203张原始光谱,其混合过程原始光谱的特征和第一批混合过程较为相似,混合初期光谱变化较为明显,随着混合的进行,光谱差异变小,光谱较为密集。[align=center][img=,488,280]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141915_02_1626619_3.png[/img][/align][align=center]图4 第二批混合过程原始光谱[/align][align=left] [b]3.2.2.2 在线混合过程结果分析[/b][/align]图5为各个光谱波段峰面积的变化图,由图中显示开始的100s内峰面积有着较大的变化幅度,随着混合的不断进行,峰面积的变化趋势不断减小并逐渐趋于稳定。[align=center][img=,516,307]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141916_01_1626619_3.png[/img][/align][align=center]图5 光谱区间峰面积图[/align][align=center](a 全光谱峰面积 bRegion 1峰面积 cRegion 2峰面积)[/align]图6为舒巴坦钠含量及标准偏差变化图,由图可知在混合的初期阶段大约0-100 s时,舒巴坦钠百分含量值及峰面积的标准偏差值有着明显的变化,全光谱峰面积的标准偏差(Full Range STD)在200-400 s间有较为明显的波段,此后随着混合过程的不断进行,四个参数变化范围均变小,模型给出的舒巴坦钠的百分含量在20%左右。由此可知前100 s是混合的主要阶段,此阶段舒巴坦钠的百分含量和标准偏差均有着明显的变化。[align=center][img=,551,327]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141917_01_1626619_3.png[/img][/align][align=center]图6 含量和标准偏差变化图[/align][align=center](a 舒巴坦钠百分含量 b 全光谱峰面积标准偏差 c Region 1峰面积标准偏差 d Region 2峰面积标准偏差)[/align]当达到混合终点时,采集表面皿底部6处的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,检测混合过程是否达到均一,表2列出来了6处的舒巴坦钠的百分含量值,由表2可知达到混合结束后得到的6处的舒巴坦钠的百分含量均在20%左右,说明混合较为均匀。同时,由于实验条件的限制加上搅拌时人为因素的影响等,各点之间含量也着较大的差异。[align=center]表2 舒巴坦钠百分含量[/align][align=center] [img=,566,84]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141918_01_1626619_3.png[/img][/align][b]3.2.3 第三批实验结果分析3.2.3.1 原始光谱图[/b]图7给出了混合过程中采集得到的207张原始光谱,由图中可知,得到的原始光谱图与第一批和第二批有着相似的结果,即混合的初期光谱差异大,因此光谱较为稀疏(偏下方的光谱),随着混合的进行,光谱间差异变小,光谱变得密集(偏上方的光谱)。[align=center][img=,505,262]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141919_01_1626619_3.png[/img][/align][align=center]图7 第三批混合过程原始光谱[/align][b]3.2.3.2 在线混合过程结果分析[/b]图8给出了混合过程中3个光谱区间峰面积的变化趋势值,由图中可知0-100s间三个光谱区间的峰面积有着明显的变化,100-200s间峰面积有着明显的变化,但是变化幅度没有前100s大,200s以后峰面积变化趋势变小。说明前200s是混合的主要阶段,峰面积变化较为明显。[align=center][img=,519,343]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141919_02_1626619_3.png[/img][/align][align=center]图 8 光谱区间峰面积图[/align][align=center](a 全光谱峰面积 bRegion 1峰面积 cRegion 2峰面积)[/align]图9为舒巴坦钠百分含量及光谱峰面积的标准偏差随时间变化的趋势图,其变化趋势和峰面积的变化趋势相似,前100s变化幅度较大,100-200s间也有较为明显的变化,但是变化幅度不是很明显,200s后舒巴坦钠的百分含量和峰面积的标准偏差均趋于稳定,说明此时光谱差异变小,混合趋于均匀。[align=center][img=,529,352]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141920_01_1626619_3.png[/img][/align][align=center]图9 含量和标准偏差变化图[/align][align=center](a舒巴坦钠百分含量变化 b全光谱峰面积标准差 c Region1峰面积标准差 d Region2峰面积标准差)[/align]表3为达到混合终点时采集表面皿底部的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱得到的不同点的舒巴坦钠的百分含量值,由表中显示6个点的舒巴坦钠的百分含量值在20%左右,但是6个点之间舒巴坦钠百分含量间存在较大的差异,测得的最小值为17.80%,其原因可能是一方面由于实验条件的限制混合不够均匀,一方面用于舒巴坦钠含量测定的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]定量分析模型也有一定的偏差,可能引起含量检测的差异存在。[align=center]表3 混合后不同点舒巴坦钠百分含量值[/align][align=center] [img=,564,66]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141921_01_1626619_3.png[/img][/align][b]3.3小结[/b]通过3个混合平行实验的进行可知所建立的基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型能够有效的监测舒巴坦钠、美洛西林钠的混合过程。由舒巴坦钠百分含量和标准偏差变化图可知两者的变化有着相关性,当舒巴坦钠的百分含量变化幅度大时,其标准偏差的变化幅度也较大,因此两者均可以用于混合过程的在线监测,证实了实验的可行性。[b]4 结论和讨论[/b]本研究采用AntarisII傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]对美洛西林钠、舒巴坦钠混合过程进行了在线监测。在研究中,分别建立了基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型,然后Antaris II傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]漫反射采样方式采集混合过程中的光谱,实时监测混合过程的进行。通过3个平行实验的在线混合过程,结果显示MBSD法和舒巴坦钠百分含量测定法均能有效的监测其混合过程,有效的证明了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于舒巴坦钠、美洛西林钠混合在线监测的可行性。此外,MBSD法因为无需进行一级数据的采集,方法较为简单且容易理解,目前常用于混合过程的在线监测。本研究中有效证实了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术在舒巴坦钠美洛西林钠样品在线混合过程中应用的可行性,在样品的在线混合监测中有着重要的应用价值和应用前景。该技术能够克服传统方法费时、繁琐等缺点,而且可以实现过程的实时在线监测,让生产者充分了解整个生产过程中的参数变化。 [b]参考文献[/b]陆婉珍, 褚小立. [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]([url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url])和过程分析技术(PAT). 现代科学仪器, 2007(004):13-17.SieslerH, Ozaki Y, Kawata S, et al. Near-infrared spectroscopy: principles .Instruments, Applications, 2002:35-181.Bhushan,K.R.,et al.Detection of breastcancer microcalcifications using a dual-modality SPECT/[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url] fluorescent probe. J Am Chem Soc, 2008. 130(52):17648-17649.贾燕花. [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术在化学药品生产过程控制应用初探. 北京协和医学院, 2011.Fevotte.G,et al.Applications of [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]spectroscopy to monitoring and analyzing the solid state during industrialcrystallization processes . Int J Pharm, 2004, 273(1):159-169.张敏.盐酸林可霉素多晶型分子构象对其红外光谱行为的影响.中国抗生素杂志, 2005, 30(009):529-532.Blanco M,R Goz"01ez Ba,E.Bertran,Monitoring powder blending in pharmaceutical processes by use of nearinfrared spectroscopy . Talanta, 2002, 56(1):203-212,田科雄.不同装载系数和混合时间对添加剂预混料混合均匀度的影响.河北畜牧兽医, 2004, 20(9):52-53.孙栋. 基于[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术的几种固体粉末混合均匀度快速检测研究. 山东大学硕士学位论文, 2012年.
呋喃西林在检测过程中添加的HCL和NAOH的作用,谢谢
做环保检测时,经常遇到安瓿瓶装的标液、盲样,打开后不易存放,请问下各路大神,哪些项目的标液、盲样,打开后可以分装到西林瓶中,有什么注意事项?总磷、汞等这些易变质、易挥发的标液是否可以分装到西林瓶?

http://ng1.17img.cn/bbsfiles/images/2012/11/201211011611_400735_2518341_3.jpg联邦制药8年7次环境违规 被曝阿莫西林爬出活虫联邦制药今年来可谓陷入多事之秋。继5月毒胶囊事件后,6月其子公司内蒙古联邦制药又被踢爆涉嫌污染环境,而昨日其位于珠海的子公司联邦制药又遭到市民投诉称,阿莫西林胶囊有效期内发现活虫,胶囊的外壳甚至已被小虫咬掉一块。这条可怜的虫子是怎么从胶囊里面爬出来的?1、是市场的监管体制不健全还是药企工作疏忽?2、毒胶囊事件还未平息,活虫又从里面爬出来了,用户该如何防范?3、药品检测标准和流程有哪些?为什么会出现如此事件?4、药监机构的发展现状如何?什么时候才能给消费者一个放心的药品?
这两天在做阿莫西林,标准上选脱掉三个水的加氢峰366作为母离子,这个没问题。sim模式366在ESI+下响应不错,但做子离子扫描时就郁闷了,裂解电压碰撞能量怎么调节都看不到二级质谱碎片,能量增加366确实被打碎了,但在50-500扫描范围就是看不见任何碎片,真是太奇怪了,它碎掉成什么东西了呢,不会都是50以下的基团吧,有没有人遇到和我相同的问题?要实在不行打算重新买标样了
新手小白,刚接触[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url],想请教走呋喃西林代谢物时,用的乙腈,0.1%甲酸水作为流动相,在4分钟左右出峰,响应值不高,离子对209-192和209-166,192碎片离子几乎是没有丰度的,而且调节它的碰撞能量也没有反应,请问这是什么原因呢?
硝基呋喃类呋喃西林响应值不好,呋喃妥因,呋喃它酮线性不好,然后唑酮没有问题,混标,不知道哪个地方出问题了,请教一下各位老师。
有谁做过阿莫西林含量和有关物质,药典中提到色谱图应与标准图谱一致,标准图谱指的是什么,哪里有,含量中提到用系统适用性对照品,哪里可以买到?
欲求购100和50mL的西林瓶,北京地区有此产品的销售商请联系我。联系方式:8072880-8313,刘先生
我们做呋喃代谢物的时候,当样品为虾时呋喃西林代谢物总是有很高的浓度,哪位高手能给解答一下。
阿莫西林有关杂质
最近做呋喃西林,曲线零点和试剂空白老是有峰还不小,但是走空瓶又没有了,排除前处理交叉污染,而且曲线线性1-20ppb的点很好,20之后的点就不成线性了很奇怪,想不出是因为啥,我要怎么排查、解决这个问题呢,在此求助各位老师。
用Elisa检测水产中四种呋喃类代谢物时,前处理方法一样,为何呋喃西林本底很高,有谁做过?怎样降本底干扰

http://ng1.17img.cn/bbsfiles/images/2012/03/201203081038_353205_1638724_3.jpg上文中的阿莫西林系统适用性对照品就是含量测定用的对照品吗?
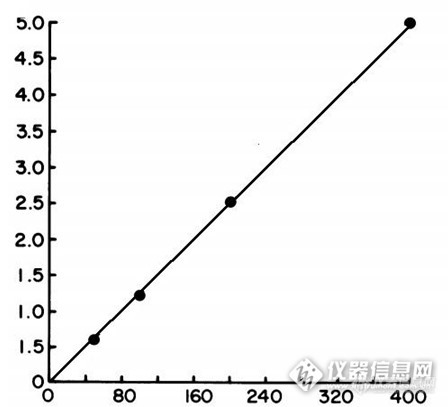
高效液相色谱法测定测定血清中替卡西林水平 卡替西林是一种半合成的抗假单胞菌青霉素,对于严重革兰阴性菌感染特别有效,除了用于治疗单胞菌感染,替卡西林也用于经验用于免疫受损的宿主。通常,这两种情况下,卡替西林总是与氨基糖苷类或头孢菌素联合应用。与青霉素联用的毒性一般是最小的,但当血清中水平高时,也会出现中枢神经系统的副作用。虽然不是常规要求,但是对于肾功能不全患者,特别与其他的β-内酰胺类抗生素联合用药时血清水平监测是很有必要的。 传统替卡西林的测定是通过微生物分析方法测定,该方法虽然划算,但这些方法总是缺乏与生化测定或免疫测定联用的特异性和精确度,而且需要最少8小时的孵育过程,不利于剂量调整。高效液相色谱法定量测定血清中替卡西林水平以及药剂中青霉素和头孢菌素和血清及尿液中替卡西林的测定在文献中均有报道,但均对于临床应用不宜,本实验做了调整优化,对于临床应用实用性较强。材料和方法: 替卡西林/替莫西林均购自药店,甲醇,氯仿,冰乙酸,盐酸,正戊醇,醋酸铵,磷酸二氢钠为分析纯或色谱纯。流动相为85(醋酸铵液):15(甲醇)醋酸铵液:醋酸铵液浓度为0.1M,并以冰醋酸调节PH为4 样品提取溶液预先配置室温保存:包含0.4N的盐酸,氯仿:正戊醇(3:1),0.1M磷酸盐缓冲液(PH=7),磷酸盐缓冲液用前按1:10用水稀释,去离子水。 标准,对照的配置:替卡西林二钠用灭菌的去离子水溶解后加入加热灭活的人血清中,配置浓度为50,100,200,400ug/ml,,并以同样方法配置250ug/ml作为对照。标准和对照血清分别以0.5ml分装,-70度保存。替莫西林以去离子水溶解于灭菌去离子水制成150ug/ml.,同法保存。标准和对照血清以及内标替莫西林用前融化。 样品制备:血清样品,标准和对照血清分别为0.35ml,加入0.15ml内标溶液,0.25ml 0.4N的盐酸,3.5ml的氯仿-正戊醇于带有螺旋盖的试管中。混合均匀后离心10分钟。上层弃去留下层。下层再加入0.35ml的磷酸盐缓冲液,混合均匀后离心10分钟。移取上层,4度保存备用。 液相条件:沃特斯2487配DAD检测器 water bondapak C18柱 (10um×4.6mm×150mm),检测波长242nm. 进样量20ml, 流速1.5ml/min 定量:标准曲线通过替卡西林的峰高与内标峰高的比率以及内标峰浓度进行绘制。 提取效率:替卡西林和内标的回收率通过比较血清提取以及相同浓素的含水制剂的峰高 精密度:日内通过向正常血清中加入替卡西林,(75ug/ml,150 ,ug/ml,300ug/ml),进行测定,日间通过三周内10次测定获得。 样品获得:该试验中应用的血清样本来自临床上那些替卡西林水平需要监测的患者。结果:1、血清中内标和替卡西林的提取后分析图谱如下:(内标和替卡西林的保留时间分别为5.4min,6.8min。http://ng1.17img.cn/bbsfiles/images/2014/10/201410300942_520789_2204138_3.png2、绝对回收率替卡西林血清回收率在29-385ug/ml范围内平均值为71%,而内标的回收率为67%,相对回收率,替卡西林在75-300ug/ml范围内平均为97%,如下图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410300943_520790_2204138_3.png3、下图为替卡西林标准曲线http://ng1.17img.cn/bbsfiles/images/2014/10/201410300943_520791_2204138_3.png讨论: 1、本实验开发了一种运用高效液相测定血液中替卡西林水平的方法,将血清加入替卡西林作为内表。采用氯仿-正戊醇进行萃取,后反萃取于磷酸盐缓冲液中。以反向C18柱,乙酸铵-甲醇水为流动相,240nm下进行检测。虽然头孢西丁,头孢噻吩,头孢呋辛等与替卡西林保留行为相似,但抗生素联合使用对于替卡西林的检测没有影响。试验表明本方法对于单用及联用抗生素时对于卡替西林的快速检测是准确,可重现的 2、本试验所采用的高效液相法分析血清中替卡西林的方法准确、重现性好,当患者联合用药时也能快速检测不干扰。 3、本试验采用内标的方法,从而克服了样品到样品间提取的变数,因为结构相似我们采用替莫西林作为内标。在提取过程和色谱行为方面也证明了采用替莫西林的可靠性。 4.该方法可用于抗生素联合用药时患者血清中替卡西林的水平测定,在患者的服用剂量调整范围内也是可适用的。

前几天碰到一个同行,聊得的这块。他告诉我,现在检测西林瓶里面有没有异物,可以用连续式灯检机。[img=,690,589]http://ng1.17img.cn/bbsfiles/images/2017/11/201711211710_01_3337770_3.png!w690x589.jpg[/img][img=,690,589]http://ng1.17img.cn/bbsfiles/images/2017/11/201711211710_01_3337770_3.png!w690x589.jpg[/img]有没有懂行的?说说,这东西怎么样啊?谢谢了。它的[url=http://www.xj-ch.com/product/97/121.html]悬臂箱[/url]系统是不是可以换别的?我不懂这个,请了解的朋友,说说。谢谢。[color=#ffffff]连续式灯检机[/color][color=#ffffff]连续式灯检机[/color]
我做的是生物制品中的氨苄西林残留,内标选的阿莫西林。仪器:美国AB3200,液相是岛津LC-20AD。流动相条件:乙腈,甲酸水(甲酸调PH3.1)梯度程序:0.01min 乙腈5%2min 乙腈5%6min 乙腈80%7min 乙腈5%8min 乙腈5%由于基质里面含有不挥发性盐,所以用了切换阀,前三分钟打进废液,后面再进质谱。这个条件一直做的很好,内标在4min出峰,氨苄西林在5.4min出峰。现在的问题是:条件不能重复了,内标峰形很怪(峰分叉,很毛糙),而且出峰时间延迟了0.5min.而氨苄西林没有变化。如果轻微变一下梯度条件,内标峰就变的很好了,所以我认为质谱是没有问题的。现在的问题就出在液相条件上,我找了很多原因,最开始换了色谱柱,换了两根(同品牌,同规格),一根还是新的,但内标出峰还是一样怪!现在就排除了柱子的问题,那么问题是不是就出在流动相条件上?!乙腈用的牌子是默克的,应该没问题吧。水是制的超纯水,以前也一直这样用的,调PH前是校正了PH仪的,PH值应该还是没问题。但问题还是没有解决!是否是梯度程序问题呢?(但以前一直都是用的这个梯度,重复性很好啊)请教高手,帮忙找下原因啊!这个问题困扰我好久了,方法学已经做完了,现在要测样品了,确出现了这种问题,小妹真的很急啊!
急急急!!!实验室检测的哌拉西林酸在水中的溶解度不合格,哪位亲有相关的文献资料送上,或者说说与那些因素有关,不胜感激







 我要推广仪器
我要推广仪器
 下载APP
下载APP