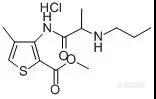
[color=black]复方盐酸阿替卡因注射液为复方制剂,是盐酸阿替卡因与肾上腺素的灭菌水溶液,作为口腔用局部麻醉剂,适用于涉及切骨术及粘膜切开的外科手术过程。[/color][color=black] [/color][img=,156,99]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211036489419_4502_2297_3.jpg!w156x99.jpg[/img][align=center][/align][align=left][b][color=black]盐酸阿替卡因(Articaine hydrochloride M.W.:320.84)[/color][/b][/align][align=center][b][color=black] [/color][/b][/align][color=black]在现有国家药品标准(YBH17082004-2015Z)分析方法中,流动相添加了离子对试剂-庚烷磺酸钠,并在pH为2.0的强酸条件下进行相应分析,不利于色谱柱的使用寿命。大曹三耀实验室参考USP方法,以冰醋酸水溶液-乙腈作为流动相,选用CAPCELL PAK C18 MGII色谱柱,实现了复方盐酸阿替卡因注射液中盐酸阿替卡因的定量和有关物质的良好分析(复方盐酸阿替卡因注射液由客户提供)。[/color][color=black]CAPCELLPAK C18 MGII[/color][color=black]液相色谱柱,其采用高纯度硅胶作为基质,通过减少硅胶微细孔的数量来增大有效比表面积;并且采用新包被技术Ultimate Polymer Coating,实现了对硅醇基极大程度的封锁,兼具分离性能和普适性能,通用性非常好。[/color][align=left][b][color=#0070c0]实验方法[/color][/b][/align][align=left][img=,500,358]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211037456969_5082_2297_3.jpg!w730x523.jpg[/img][/align][align=left]图1[color=black]盐酸阿替卡因[/color]对照品及供试品溶液[/align][align=left][img=,500,248]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211038541919_2603_2297_3.jpg!w572x284.jpg[/img][/align][align=center][/align][align=center][/align][color=black]为进行有关物质分析,该实验将注射液样品以流动相稀释100倍,作为有关物质供试品溶液,再将该有关物质供试品溶液以流动相进一步稀释100倍,作为自身对照溶液。以冰醋酸水溶液-乙腈作为流动相,选用CAPCELL PAK C18 MGII色谱柱,通过调整流动相比例及柱温,最终在18%乙腈、柱温30℃条件下实现了盐酸阿替卡因供试品溶液及对照品的良好分析。[/color]如图2、3,使用CAPCELL PAK C18 MGII色谱柱进行分析,盐酸阿替卡因和有关物质均能得到良好分析结果,主峰与峰前杂质得到了良好分离,分离度为1.90(见表1)[img=,400,311]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211045536855_9516_2297_3.jpg!w574x447.jpg[/img][img=,400,295]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211045540875_8483_2297_3.jpg!w698x516.jpg[/img][align=left] 图2 [color=black]盐酸阿替卡因[/color]有关物质供试品溶液及空白 图3 自身对照溶液[/align][align=center][/align][align=left]表1 有关物质结果详表[/align][align=left][img=,600,323]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211042513275_6690_2297_3.jpg!w786x424.jpg[/img][/align][align=center][/align]综上实验结果,使用CAPCELL PAK C18 MGII S5 4.6mm i.d.×250 mm色谱柱,以冰醋酸水溶液-乙腈为流动相体系,在30°C柱温条件下,能够实现复方盐酸阿替卡因注射液中盐酸阿替卡因的定量和有关物质的良好分析。[color=black] [/color]
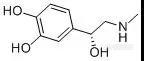
复方盐酸阿替卡因注射液为复方制剂,是盐酸阿替卡因与肾上腺素的灭菌水溶液,作为口腔用局部麻醉剂,适用于涉及切骨术及粘膜切开的外科手术过程。[img=,144,61]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191545418661_4518_2222981_3.jpg!w144x61.jpg[/img][color=black] [/color][color=#3e3e3e]肾上腺素 L(-)-Epinephrine M.W. : 183.2 [/color][b]在国家药品标准(YBH17082004-2015Z)[/b]中,在对复方盐酸阿替卡因注射液中肾上腺素进行分析时,使用[b]甲醇-水[/b]进行梯度洗脱,但由于[b]肾上腺素极性较强[/b],即使初始梯度为纯水相条件,肾上腺素仍紧邻死时间出峰,[b]保留不佳,易受到溶剂峰干扰,无法进行准确定量。[/b]我们分别尝试使用反相柱CAPCELL PAK C18 MGII加离子对试剂,以及直接使用离子交换色谱柱CAPCELL PAK SCX UG80两种方式,对复方盐酸阿替卡因注射液中肾上腺素和硫酸肾上腺素进行保留分析(复方盐酸阿替卡因注射液由客户提供)。CAPCELL PAK C18 MGII液相色谱柱,其采用高纯度硅胶作为基质,通过减少硅胶微细孔的数量来增大有效比表面积;并且采用新包被技术Ultimate Polymer Coating,实现了对硅醇基极大程度的封锁,兼具分离性能和普适性能,通用性非常好。CAPCELL PAK SCX UG80是强阳离子交换柱,使用高纯度硅胶,填料中金属杂质很少,使配位化合物的吸附得到了极大程度抑制,兼具聚合物和硅胶填料的优点。[b][color=#0070c0]实验方法[/color][color=#0070c0]方法一[/color][color=#0070c0]使用[/color][color=#0070c0]CAPCELL PAK C18MGII[/color][color=#0070c0]色谱柱[/color][color=#0070c0]+[/color][color=#0070c0]离子对试剂[/color][/b]如图1,对肾上腺素对照品溶液进行分析,肾上腺素主峰保留时间为5.69 min,拖尾因子为1.19,理论塔板数为12538。在相同色谱条件下,尝试对亚硫酸肾上腺素标准品及注射液中的亚硫酸肾上腺素进行分析。如图3,亚硫酸肾上腺素标准品溶液能够得到良好分析结果,注射液(客户提供的样品)中未明显见亚硫酸肾上腺素出峰,保留时间为3.55min,拖尾因子为1.14,理论塔板数为14955。[b][color=#0070c0]方法二[/color][color=#0070c0] CAPCELL PAK SCX UG80[/color][color=#0070c0]色谱柱[/color][/b][color=#000000]考虑到使用离子对试剂的流动相条件具有流动相配制麻烦、有损色谱柱寿命、平衡时间长等缺点,我们也尝试使用键合磺酸基团的强阳离子交换柱 ——CAPCELL PAK SCX UG80进行分析。[/color][color=#000000]如图4,在流动相中添加磷酸二氢铵,通过对盐浓度进行调整,在5 mmol/L磷酸二氢铵(磷酸调pH=2.5)条件下,亚硫酸肾上腺素保留时间为3.32 min,然而出现峰形拖尾现象,拖尾因子为2.0,不如CAPCELL PAK C18 MGII色谱柱添加离子对试剂所得分析结果好。[/color][align=center][/align][align=left][img=,400,284]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191547381421_926_2222981_3.jpg!w584x416.jpg[/img] [img=,400,276]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191548310561_8067_2222981_3.jpg!w572x395.jpg[/img][/align][align=left][img=,400,166]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191547576748_522_2222981_3.jpg!w612x254.jpg[/img] [img=,400,167]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191548525761_4184_2222981_3.jpg!w624x262.jpg[/img][/align][align=left]图1 MGII分析肾上腺素对照品溶液结果(离子对条件) 图2 MGII分析注射液结果(离子对条件)[/align][align=center][/align][img=,400,258]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191550354951_2539_2222981_3.jpg!w644x416.jpg[/img] [img=,400,250]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191555486850_3396_2222981_3.jpg!w644x403.jpg[/img][img=,400,147]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191551260881_9828_2222981_3.jpg!w696x256.jpg[/img] [img=,400,164]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191556163846_9739_2222981_3.jpg!w632x260.jpg[/img][align=left]图3 MGII分析亚硫酸肾上腺素标准品和注射液结果(离子对条件) 图4 SCX UG80分析亚硫酸肾上腺素对照品溶液和供试品溶液[/align][align=left][/align][align=left]综上实验结果,使用中等极性色谱柱CAPCELL PAK C18 MGII S5 4.6 mm i.d. × 250 mm,在流动相中添加5 mM辛烷磺酸钠、30°C柱温条件下进行梯度洗脱,能够实现复方盐酸阿替卡因注射液中肾上腺素和亚硫酸肾上腺素的良好保留与分析。[/align][b][color=#0070c0][/color][/b][align=left][b][color=#0070c0] [/color][color=#0070c0] [/color][/b][/align]
各位师兄师姐,有没有拿二级测氨基糖苷类(我做庆大霉素、依替米星和阿米卡星)吗?柱子和流动相用的什么呀?七氟丁酸实验室不给用,HPLC实验室只有紫外和荧光检测器,不能做ELSD和FID。我的课题刚上手,好多不太懂,老师催着摸方法,好纠结。。。谢谢各位师兄师姐~
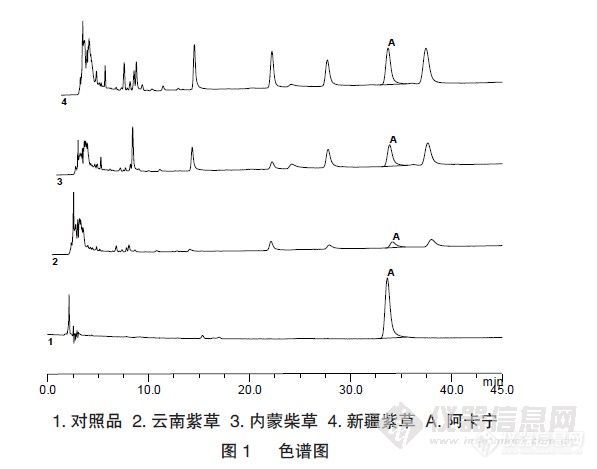
【作者】 谢清春; 陈燕忠; 吕竹芬;【Author】 XIE Qingchun CHEN Yanzhong LV ZhufenInstitute of Material Medica, Guangdong Pharmaceutical College, Guangzhou 510006, Chin【机构】 广东药学院药物研究所;【摘要】 目的测定不同产地的紫草中β,β’-二甲基丙烯酰阿卡宁(以下简称阿卡宁)的含量,比较不同溶剂对紫草中阿卡宁的提取率,并对阿卡宁的乙醇溶液稳定性进行考察。方法采用高效液相色谱法测定阿卡宁的含量,色谱柱为Dikma Diamonsil C18柱(200mm×4.6mm,5μm),以乙腈-水-甲酸(700:300:0.5)为流动相,流速1mL·min-1,检测波长275nm。测定了内蒙紫草、新疆紫草和云南紫草中阿卡宁的含量,考察了75%乙醇、乙醇、石油醚和液体石蜡对紫草中阿卡宁的提取率;将阿卡宁0.1mg·mL-1乙醇溶液于冰箱4℃存放,定期进样测定,并与新配制时测定的峰面积比较。结果测定的三种紫草中,以新疆紫草含量最高;乙醇和石油醚对阿卡宁的提取率高而75%乙醇和液体石蜡的提取率低;阿卡宁的乙醇溶液随存放时间的增加而峰面积变小。结论提取溶剂对紫草中阿卡宁提取率影响较大;由于阿卡宁溶液的不稳定性,选择阿卡宁作为评价含紫草药材制剂的指标成分并不合适。 更多还原【Abstract】 Objective To determine the β,β’-dimethylacrylalkannin in lithospermum erythrorhizon, compare the extractingrate of different solvents to β,β’-dimethylacrylalkannin and study the stability of standard β,β’-dimethylacrylalkanninsolution of alcohol. Methods A HPLC method was used to determine the β,β’-dimethylacrylalkannin. Dikma Diamonsil C18chromatogram column(200 mm×4.6 mm, 5μm) was used, Acetonitrile-water-Formic acid(700:300:0.5) as mobile phase, flowrate at 1mL·min-1, detection wavelength at ... 更多还原【关键词】 紫草; β,β’-二甲基丙烯酰阿卡宁; 提取率; 高效液相色语法; 稳定性; 【Key words】 Lithospermum erythrorhizon; β,β’- dimethylacrylalkannin; Extracting rate; HPLC; Stability; http://ng1.17img.cn/bbsfiles/images/2012/08/201208201119_384576_2352694_3.jpg
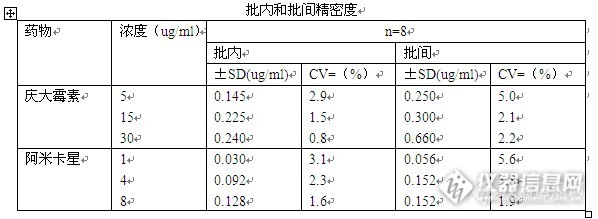
高效液相色谱法快速测定血清中的庆大霉素和阿米卡星庆大霉素和阿米卡星是用于严重革兰氏阴性细菌感染治疗的氨基糖苷类抗生素。像其他氨基糖苷类抗生素一样,庆大霉素和阿米卡星的有效治疗范围狭窄容易导致肾脏毒性和耳毒性。因此,监测庆大霉素和阿米卡星血清中的水平对于安全和有效的临床应用十分重要。最近,高压液相色谱法已被报道用于氨基糖苷类抗生素的测定。多数测定需要柱前或柱后衍生荧光检测。这些方法涉及耗时的预处理,如血清中氨基糖苷类抗生素的柱萃取以及溶剂。本实验开发了一种较为简单的预处理方法来测定血清中的庆大霉素和阿米卡星,并与荧光偏振免疫测定法进行了比较。材料和方法:庆大霉素和阿米卡星购自药店,邻苯二甲醛(瑞尔丰化工),2-巯基乙醇、庚烷磺酸钠、1,2-乙烷二磺酸二钠、色谱乙腈、蒸馏水。蛋白质沉淀剂(10mM辛烷磺酸钠溶解于3.5%高氯酸中)。庆大霉素的流动相为含有22 mM的1,2 - 乙烷二磺酸二钠和5mM辛烷磺酸钠的水 - 乙腈混合物(80:20,体积/体积),用乙酸调节pH至约3.5。阿米卡星的流动相为含有37 mM的1,2 - 乙烷二磺酸二钠和5mM辛烷磺酸钠的水 - 乙腈混合物(80:20,体积/体积),用乙酸调节pH至约3.5。仪器和色谱条件:安捷伦高效液相色谱仪1200,UV检测器,色谱柱:安捷伦Agilent液相色谱柱4.6﹡150﹡5u,流动相的流速保持在1毫升/分钟。邻苯二甲醛试剂用泵以0.6毫升/分钟的流速。实验部分:将50ul血浆加入到50ul蛋白质沉淀剂,涡旋混合数秒后,使用KM-15200离心机离心2分钟(15000rpm),离心后上清液进样。正常血清,分别加入已知量的庆大霉素和阿米卡星,进行分析,根据加入量和峰面积绘制标准曲线。荧光偏振免疫通过市售的试剂盒进行。结果:图1为庆大霉素的标准色谱图,庆大霉素加入对照血清的色谱图以及对照血清的色谱图。(其中交标庆大霉素的含量为15ug/ml)http://ng1.17img.cn/bbsfiles/images/2014/09/201409300949_516466_2188679_3.jpg图2为阿米卡星的标准色谱图,阿米卡星加入对照血清的色谱图以及对照血清的色谱图,经过蛋白质沉淀剂处理后的血清和未处理的色谱图基本形同,皆未见到影响抗生素检测的干扰杂质峰。http://ng1.17img.cn/bbsfiles/images/2014/09/201409300949_516467_2188679_3.jpg以峰面积作为纵坐标,血清中的药物浓度为横坐标得到庆大霉素的线性标准曲线(3-50ug/ml)为y =0.979x - 0.006,阿米卡星的线性标准曲线(0.5-10ug/ml)为y= 0.998x - 0.04根据庆大霉素和阿米卡星在血清中不同浓度的重复测定,得到庆大霉素和阿米卡星的批内变异系数分别为0.8 -2.9%和1.6-3.1%,批间变异系数为2.0-5.0%及1.9-5.6%。http://ng1.17img.cn/bbsfiles/images/2014/09/201409300949_516468_2188679_3.jpg回收率试验:分别将15ug/ml的庆大霉素水溶液加入含有15ug/ml阿米卡星的血清中和将4ug/ml的阿米卡星水溶液加入4ug/ml的血清中。将它们的峰面积进行比较。基于三个样品的平均值,庆大霉素和阿米卡星的分别为99.0%和98.5%.通过该方法获得的结果与用荧光偏振免疫测定法进行了比较,回归方程和相关系数分别为庆大霉素:y=1.027x - 1.090,n =44和r=0.996。阿米卡星y=0.998x - 0.206, n =42和r=0.957。讨论:本实验采用了蛋白质沉淀剂,排除干扰物,再将样品进行分析,优化的分析方法与荧光偏振免疫测定法进行了比较种,该方法方法的优点是速度快,操作简便,重复性好。因此,该方法可用于该药物的分析。
在下现在做阿卡波糖溶出的液相检测,根据新药转正标准的色谱条件,采用C18柱,流动相磷酸盐缓冲溶液:乙腈=95:5 主峰总是分叉,求高手指点!
各位大神哪位有固体咖啡因的COA单子,扫描件即可,最近在做仪器的PQ用到咖啡因,但自己手上的咖啡因coa单子没有了,求救各位了,很急!!
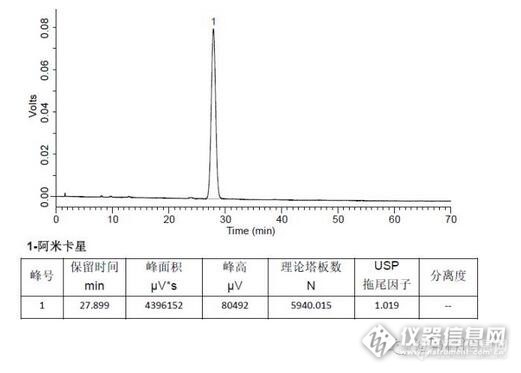
1 样品制备【1】含量测定供试品溶液:取样品适量,用流动相稀释制成每毫升2.5mg阿米卡星。【2】有关物质2.1 供试品溶液:取样品适量,用流动相A稀释制成每毫升5mg阿米卡星。2.2 对照品溶液:取供试品用流动相A稀释成每毫升含阿米卡星50μg的溶液。2 分析条件【1】分析条件(有关物质)http://ng1.17img.cn/bbsfiles/images/2016/03/201603281649_588463_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/03/201603281649_588464_1610895_3.jpg【2】分析条件(含量测定)http://ng1.17img.cn/bbsfiles/images/2016/03/201603281650_588465_1610895_3.jpg3 色谱图I 客户A检测方案有关物质供试品:http://ng1.17img.cn/bbsfiles/images/2016/03/201603281650_588466_1610895_3.jpg有关物质对照品:http://ng1.17img.cn/bbsfiles/images/2016/03/201603281650_588467_1610895_3.jpg含量测定供试品:http://ng1.17img.cn/bbsfiles/images/2016/03/201603281651_588468_1610895_3.jpgⅡ客户B检测方案有关物质供试品:http://ng1.17img.cn/bbsfiles/images/2016/03/201603281651_588469_1610895_3.jpg有关物质对照品:http://ng1.17img.cn/bbsfiles/images/2016/03/201603281651_588470_1610895_3.jpg含量测定供试品:http://ng1.17img.cn/bbsfiles/images/2016/03/201603281652_588471_1610895_3.jpg
阿卡波糖醋酸盐PH=4.5溶出介质有干扰,主峰和介质峰出风时间在一起,而且介质峰包着主峰,这该怎么解决,流动相比例调过好多次都无法改变,请高手指点
[align=center]阿卡波糖系统适用性试验[/align][align=center]-遵循2015中国药典[/align][align=center] [/align][b]色谱条件:[/b]色谱柱:Kromasil 100- 5-NH2,4.6*250mm货号:M05NHA25流动相:磷酸盐缓冲液:乙腈=25:75流速:2ml/min柱温:室温波长:210nm进样量:10uL[b]系统适用性实验图谱:[/b][align=center][img=,554,268]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181713142962_1500_2785_3.png[/img][/align][b]结论[/b][list=1][*]杂质I相对于阿卡波糖的保留时间约为0.9[*]杂质I的峰高与杂质I和阿卡波糖两峰之间的峰谷之比大于2.0[*]理论塔板数按阿卡波糖计算大于2000[/list]结果完全符合2015中国药典 [b]小贴士:[/b]实验前不要忘记平衡色谱柱和柱效测试哦[img]http://simg.instrument.com.cn/bbs/images/default/em09502.gif[/img]
海洛因、咖啡因的FTIR检验及谱图解释 摘要: 目的提高用红外光谱法鉴定海洛因、咖啡因纯品、混合物伪品的水平。方法用红外光谱法有针对性地选择特征峰,探明海洛因、咖啡因红外光谱与结构的关系。结果获得海洛因、咖啡因的特征峰。结论该方法克服了鉴定中的盲目性。 关键词:海洛因;咖啡因;红外光谱 Analysis of heroin and caffeine by FTIR and interpretations of the IR spectra FENGji-min ,LIU Shi-hai ABSTRACT: Objective To enhanced the capability to identify and differentiate the pure/mixture as well as some fakes heroin and caffeine.Method Explain away relationship between the spectra with structure characteristics of heroin and caffeine.Result Characteristic peaks.for beroin and caffeine were selected、Conclusion Pure/mixture as well as some fakes heroin and caffeine can be identified accurately with FTIR. KEY WORDS: heroin;caffeine;infrared spectroscopy 红外光谱法是国际上常用的毒品检验方法。红外光谱分析毒品主要有以下几个特点:所需检材量小,一般只需微克级;不破坏检材,样品仍可进行其它分析;操作简便,不需要特殊的前处理,10分钟可以完成一个样品分析;不需要其它试剂,不污染环境,有益于操作人员健康;重现性好,特异性强;适应性广,受杂质种类限制少。 目前有关红外光谱检验毒品的文献,大都只介绍了各种毒品纯品的红外光谱,很少对其吸收带做必要的解释。而不了解各吸收带与结构的对应关系做毒品鉴定,带有极大的盲目性,特别是分析混有杂质的毒品,无论是计算机检索,还是人工识图,都十分困难。鉴于目前这种情况,笔者在大量实验的基础上对海洛因盐酸盐和咖啡因的红外光谱解释做了初步尝试,以期与国内同行进行交流。 1 海洛因纯品的红外光谱及其解释 海洛因(heroin),是吗啡的衍生物,其盐酸盐的化学结构式见图。 海洛因盐酸盐的红外光谱中3439/cm为N-H的伸缩振动吸收,2957/cm为3个CH3中C-H反对称伸缩振动的偶合,2632、2524、2083/cm 是海洛因盐酸盐中叔胺盐离子的N+-H对称和反对称伸缩振动吸收。1762/cm为与苯环相连的乙酰氧基的羰基的伸缩振动,1738/cm为与6元环相连的乙酰氧基的羰基的伸缩振动吸收。二个羰基的伸缩振动之所以峰位有差别,是因为苯环的电负性比6元环的吸电性强,屏蔽作用大。吸电子的诱导效应使成键的电子密度向键的几何中心接近,降低了羰基C=0的极性,增加了双键性,伸缩振动力常数增加。吸电子基团的诱导效应和苯环上大的取代基的屏蔽作用导致羰基伸缩振动更多的向高频位移。1242和1037/cm两条谱带分别归属于=C-O-C=0中O-C=0和-O-C=的反对称和对称伸缩振动,是乙酰氧基光谱中两条最特征的谱带。两者和1761、1737/cm谱带结合起来指示酯类的存在。1630/cm为六元环上C=C的伸缩振动,乙酰氧基的强吸电性使其强度增大。1492、1445/cm是苯环的骨架振动。1445/cm也包含=N-CH3中C-H的变形振动吸收。 1469、1369/cm谱带分别为甲基的反对称变角振动和对称变角振动。它们的强度明显增强,是由于甲基直接和羰基相连所引起的,是乙酰结构的明显特征。1180、1157、1131为C-O的反对称伸缩振动和对称伸缩振动。1106/cm为同时与苯环和6元环相连的C-O-C键的伸缩振动。911/cm属于六元环上-CH=CH-中反式CH面外弯曲振动。764、696/cm为苯环上2个相邻氢原子的面外变角振动吸收。 2 咖啡因纯品的红外光谱及其解释咖啡因(caffeine)又称咖啡碱。由茶叶或咖啡中提得的一种生物碱。也可人工合成。呈白色晶体或粉末。密度1.23。熔点234-237℃。小剂量作用于大脑皮层高位部分,促使精神兴奋,较大剂量可直接兴奋呼吸中枢和血管运动中枢。1996年1月国家卫生部将其列入第一类精神药品。 1695/cm是2位碳原子上羰基C=O的伸缩振动,由于受邻位N原子诱导效应的影响所以出现在较高的波数。1659/cm是6位碳原子上C=O的伸缩振动,它既受邻位N原子诱导效应的影响,也与邻位4、5原子间的双键形成共轭。共轭使C=O双键特性减弱,力常数降低,伸缩振动向低频位移,同时强度增加。3114/cm是芳香环C-H的伸缩振动,其变角振动出现在745/cm。4、5原子C=C伸缩振动和8、9原子C=N伸缩振动的叠加出现在1600/cm和1551/cm。N-CH3的C-H伸缩振动出现在2959/cm,CH3不仅受N原子的影响,而且N原子也受邻近原子的影响。3个N-CH3的N原子与不同的原子相连,C-H变角振动出现在多个位置,它们分别是1485、1456、1426、1404/cmo O=C-N 中C-N的伸缩振动分别出现在1551和1360/cm。N3-C4的伸缩振动出现在1026/cm。1074/cm为C5-C6的伸缩振动。 3 海洛因和咖啡因混合物的检验 从毒贩手中缴获的海洛因等毒品中经常掺杂有咖啡因。例如从云南发生的一起贩毒大案中缴获的毒品(公安部物证鉴定中心编为滇111号),经红外光谱检验,其红外光谱如图。把它与前面两图相比较可以发现:3116、2957、1705、1660、1549、1284、1241、974、761、745、611、480、447、426/cm为咖啡因的红外吸收;3437、2957、2634、2088、1762、1737、1489、1445 1366 1241 1182 1156 1132、1109 1033、974、912、873、837、798、761、698、644、522/cm为海洛因盐酸盐的红外吸收。经进一步检验得知,该样品中海洛因盐酸盐与咖啡因的质量比为1:0.8。 样品2则是上海一起贩毒大案中缴获的毒品(公安部物证鉴定中心编为沪85号),经红外光谱检验,其红外光谱图与前两图相比较可以发现 3113、2955、1696、1658、1549、1449、1287、1241975、748、446、423/cm为咖啡因的红外吸收。3446、2955 2634 2086 1763 1735 1487、1241 1106、1037、975、910、873、798、696、571、472/cm为海洛因盐酸盐的红外吸收。
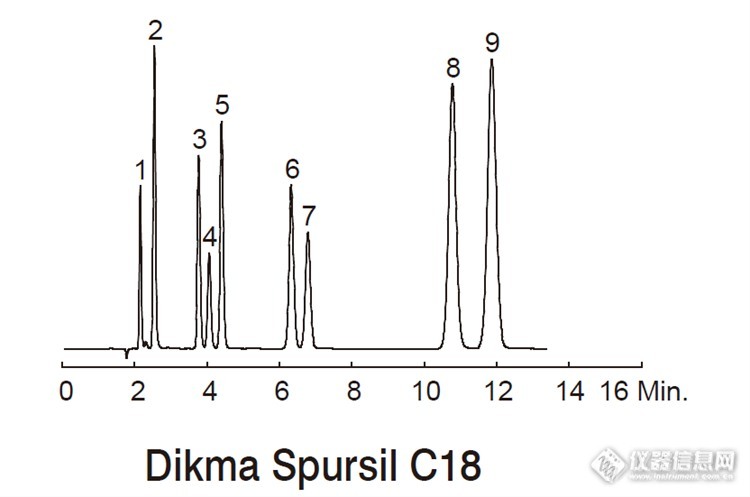
咖啡的主要成分是咖啡因,可以作用于神经细胞中一种叫做腺嘌呤核苷的化学物质,是一种中枢神经兴奋剂,能够暂时的驱走睡意并恢复精力。 不过,咖啡对有些人是有好处的,但是对某些人却产生负面影响,这主要是与咖啡因的在不同人的代谢能力有关的。 咖啡因在肝脏中被分解产生三个初级代谢产物副黄嘌呤,可可碱,茶碱。咖啡因在摄取后45分钟内被胃和小肠完全吸收。吸收后它会分布于身体的所有器官之中,转化过程符合化学动力学一级反应,这些化合物进一步代谢,最终通过尿液排泄。 如果某些人的这个酶的代谢比较快,摄入的咖啡因很快就会被清除出体外,因此咖啡因起作用的效果就很有限,不能令人产生特别明显的兴奋感。而对于另一些人,他们这个酶代谢速度慢,咖啡因在体内的清除速度很慢,起作用时间也就较长,这样的人往往一杯咖啡就会令他们夜不能寐,有的还会影响食欲,呕吐和痉挛,也可能出现胃炎或心脏病等不良反应。 所以这也解释了一般人普遍担心的咖啡会影响睡眠问题,其实和你对咖啡因的代谢力有很大的关系。由于每个人对咖啡因代谢的能力不同,平均来说,咖啡因在体内的运作,大约能维持3~4个小时,所以即使在晚餐后饮用,也不至于造成太大的困扰。但有些人的代谢力较差,可能会持续作用8~12个小时;或者体质对咖啡因比较敏感,就得特别注意喝咖啡的时间,避免影响作息,因为不论是多喝开水或是增加运动,都无法有效的促使咖啡因快速代谢。*********************************** 以上对咖啡因的代谢方面做了简单的科普,可如何对咖啡因代谢物进行检测呢?由于咖啡因代谢物从化学结构上来看,这一类化合物具有相似的母体结构,不同之处在于甲基的位置,属于位置异构体。(见下图)http://ng1.17img.cn/bbsfiles/images/2016/01/201601071109_581149_2452211_3.png 对于分离这些代谢物来说,对色谱柱的分离能力要求比较高,下面我们看看使用迪马Spursil色谱柱分离9种咖啡因代谢物的分离效果情况色谱柱:Spursil C18规格:150 x 4.6 mm, 5 μm流动相:甲醇/ 水+1% 乙酸=10/90流速:1.0 mL/min柱温:室温检测器:UV 254 nm样品:1. 尿酸2. 黄嘌呤3. 7- 甲基黄嘌呤4. 1- 甲基尿酸5. 3- 甲基黄嘌呤6. 1,3- 二甲基尿酸7. 可可碱8. 1,7- 二甲基黄嘌呤9. 茶碱http://ng1.17img.cn/bbsfiles/images/2016/01/201601071110_581151_2452211_3.png总结:Spursil 色谱柱能够在13分钟之内将它们全部分开且达到基线分离~棒棒哒http://simg.instrument.com.cn/bbs/images/default/em09505.gif
咖啡因的特性能够提升大脑和身体的能力。1979年,研究人员对比了摄取咖啡因的运动运与未摄取的运动员在长距离自行车项目中的表现,发现摄入咖啡因的运动员表现增加7%。其它也有很多研究证实了咖啡因对人体机能有着短时的提升作用。咖啡因也能与其它药结合提高药效。咖啡因能使头痛药的功效提高40%,并能提高身体吸收药品的速度,缩短药品起作用的时间。所以,在很多药品成分中都会有咖啡因。鉴于咖啡因的特性及作用,人们会对其有一定的依赖性。但由于个体差异,每个人可以摄入的咖啡因的量是不同的,这是由人体基因决定的。对于想知道自己一次摄入多少咖啡因才是最合适的人来说,要不赶紧做个咖啡因代谢基因检测了解一下~有咖啡因成分的咖啡、茶、软饮料及能量饮料十分畅销各类饮品中咖啡因含量对比表【咖啡】 咖啡因含量 80mg(一般含量范围 40-170mg)【红茶】 咖啡因含量 40mg(一般含量范围 25-110mg)【乌龙】 咖啡因含量 30mg(一般含量范围 12-55mg)【绿茶】 咖啡因含量 20mg(一般含量范围 8-30mg)【普洱】 咖啡因含量 2mg (含量极低)【麦茶】 咖啡因含量 0mg【可乐】 咖啡因含量 50mg(一般含量范围 40-80mg)【红牛】 咖啡因含量 80mg(一般含量范围 80-100mg)
液相色谱校验用的咖啡因哪里能买到,或者有什么代替品吗,最近要做校验,咖啡因管制了买不到。
阿咖酚散流动相,取量,比例的方面
咖啡因的特性能够提升大脑和身体的能力。1979年,研究人员对比了摄取咖啡因的运动运与未摄取的运动员在长距离自行车项目中的表现,发现摄入咖啡因的运动员表现增加7%。其它也有很多研究证实了咖啡因对人体机能有着短时的提升作用。咖啡因也能与其它药结合提高药效。咖啡因能使头痛药的功效提高40%,并能提高身体吸收药品的速度,缩短药品起作用的时间。所以,在很多药品成分中都会有咖啡因。鉴于咖啡因的特性及作用,人们会对其有一定的依赖性。但由于个体差异,每个人可以摄入的咖啡因的量是不同的,这是由人体基因决定的。对于想知道自己一次摄入多少咖啡因才是最合适的人来说,要不赶紧做个咖啡因代谢基因检测了解一下~有咖啡因成分的咖啡、茶、软饮料及能量饮料十分畅销各类饮品中咖啡因含量对比表【咖啡】 咖啡因含量 80mg(一般含量范围 40-170mg)【红茶】 咖啡因含量 40mg(一般含量范围 25-110mg)【乌龙】 咖啡因含量 30mg(一般含量范围 12-55mg)【绿茶】 咖啡因含量 20mg(一般含量范围 8-30mg)【普洱】 咖啡因含量 2mg (含量极低)【麦茶】 咖啡因含量 0mg【可乐】 咖啡因含量 50mg(一般含量范围 40-80mg)【红牛】 咖啡因含量 80mg(一般含量范围 80-100mg)
咖啡因的特性能够提升大脑和身体的能力。1979年,研究人员对比了摄取咖啡因的运动运与未摄取的运动员在长距离自行车项目中的表现,发现摄入咖啡因的运动员表现增加7%。其它也有很多研究证实了咖啡因对人体机能有着短时的提升作用。咖啡因也能与其它药结合提高药效。咖啡因能使头痛药的功效提高40%,并能提高身体吸收药品的速度,缩短药品起作用的时间。所以,在很多药品成分中都会有咖啡因。鉴于咖啡因的特性及作用,人们会对其有一定的依赖性。但由于个体差异,每个人可以摄入的咖啡因的量是不同的,这是由人体基因决定的。对于想知道自己一次摄入多少咖啡因才是最合适的人来说,要不赶紧做个咖啡因代谢基因检测了解一下~有咖啡因成分的咖啡、茶、软饮料及能量饮料十分畅销各类饮品中咖啡因含量对比表【咖啡】 咖啡因含量 80mg(一般含量范围 40-170mg)【红茶】 咖啡因含量 40mg(一般含量范围 25-110mg)【乌龙】 咖啡因含量 30mg(一般含量范围 12-55mg)【绿茶】 咖啡因含量 20mg(一般含量范围 8-30mg)【普洱】 咖啡因含量 2mg (含量极低)【麦茶】 咖啡因含量 0mg【可乐】 咖啡因含量 50mg(一般含量范围 40-80mg)【红牛】 咖啡因含量 80mg(一般含量范围 80-100mg)
各种饮料、药品等都含有咖啡因,所以有时出现不经意之间过量摄取的可能。为了避免咖啡因中毒风险应注意哪些问题呢?日本药科大学校长进行如下解答。首先除了咖啡、茶等饮料之外,还有很多食品或药品也含有咖啡因。众所周知咖啡豆、茶叶、可可豆等都含有咖啡因,以此为原料做成的咖啡、绿茶、红茶、巧克力、可可饮料也同样含有咖啡因。而且咖啡或茶叶中提取的咖啡因精制之后添加到医药品中,作为治疗头痛的药品、感冒药、驱赶困倦的药物使用。咖啡因还可以作为食品添加剂使用,如为了给食品附加苦味添加到可乐等清凉饮料或保健食品中。咖啡因对人体心血管和中枢神经起刺激作用,适量摄取时可提高人体活跃度。但短时间内大量摄取时会出现头痛、眩晕、胃痉挛、恶心、呕吐等急性症状。过量摄取导致咖啡因中毒时会出现无法入睡、心悸等全身症状,甚至可导致死亡。咖啡因对人体有一定的风险,但日本至今还未制定一日允许摄取量。参考其他国家的数据,健康成年人的每日推定允许摄取量约为400毫克,换算为咖啡大约3大杯,其他多数饮料中也含有咖啡因,所以饮用时一定确认所含有的咖啡因含量,注意不要过量摄取。而且作为治疗头痛药使用的咖啡因片,每片含有咖啡因100~300毫克。某些健康辅助食品中也含有高浓度的咖啡因。所以服用这些药品的同时饮用含有咖啡因饮料其风险非常大,所以服用药品时一定要控制含咖啡因饮料的摄取。还要注意咖啡因与中成药的协同作用。中成药中的麻黄所含有的麻黄碱与咖啡因合用时有相乘的功效,不知不觉中过量摄取时会出现心脉跳动增加、心悸,血压升高等症状。哮喘时使用的支气管扩张药物中也含有麻黄碱,所以服用这类药物时一定要控制饮用含有咖啡因饮料。(文章来源:食品伙伴网)
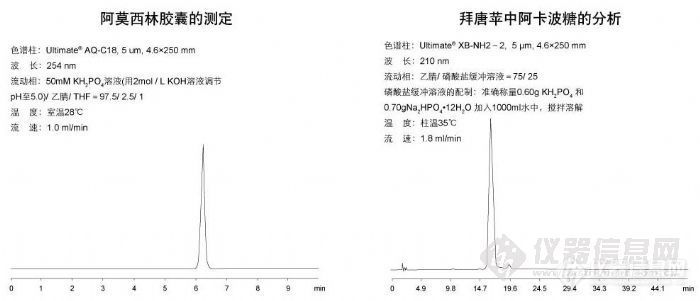
阿莫西林胶囊的测定和拜唐平中阿卡波糖的分析http://ng1.17img.cn/bbsfiles/images/2009/10/200910311058_179313_1896702_3.jpg

作者:李昂; 胡翮; 郭丙炎;(湖南省湘潭市药品检验所;)摘要:目的用反相高效液相色谱(RP-HPLC)法测定盐酸丁卡因的含量。方法色谱柱为Diamonsil C18柱(150mm×4.6mm,5μm),流动相为水-甲醇-乙腈(45∶45∶10,v/v,含0.02%庚烷磺酸钠,0.34%磷酸二氢钾,三乙胺调pH至7.0),流速为1.0mL/min,检测波长为314nm,柱温为室温。结果盐酸丁卡因质量浓度在5.034~161.1μg/mL范围内与峰面积线性关系良好,回归方程为Y=5.6572×105X-9.2039×105(r=0.9999),平均回收率为100.76%,RSD为0.41%(n=6)。结论RP-HPLC法简便、可靠、准确,可作为盐酸丁卡因注射液的含量测定方法。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208271103_386360_1606903_3.jpg
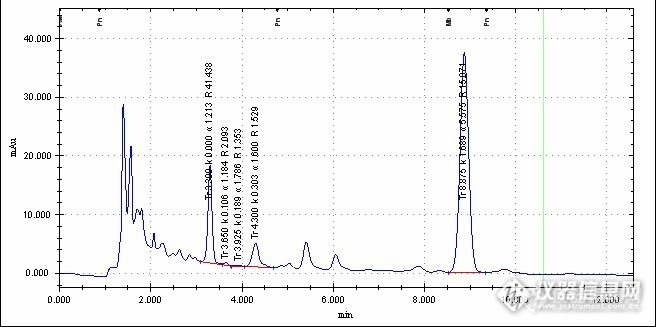
锁阳咖啡中咖啡因含量的测定锁阳咖啡以天然锁阳为基础,配以速溶咖啡,是近几年涌现出的一种新型的固体饮料。锁阳主产于甘肃、青海、内蒙,其中以甘肃嘉酒地区的锁阳产量最大、质地最优。锁阳可调节生理机能、均衡营养、促进血液循环、滋肝健肾。锁阳咖啡是锁阳和咖啡的有机结合,越来越受到消费者的青睐。但是人们在饮用锁阳咖啡的同时却容易忽略咖啡的品质,咖啡因是咖啡中的一种重要成分,也是衡量其质量的一项重要指标,作为一种中枢兴奋剂,能兴奋大脑皮层,但易上瘾,因而国家对其制定了相应的标准。本实验用SN/T 1391-2004 《进出口速溶咖啡检验规程》中速溶咖啡中咖啡因的测定方法测定锁阳咖啡中咖啡因的含量。http://ng1.17img.cn/bbsfiles/images/2013/07/201307291536_454526_2764104_3.jpg1.方法提要样品用水溶解过滤后,用配有紫外检测器的高效液相色谱(HPLC)测定咖啡因,外标法定量。2.试剂和材料所有试剂除特殊注明外,均为分析纯,水为超纯水。2.1 乙睛:色谱纯;2.2 咖啡因标准品:纯度≥99%;2.3 标准储备液(100m g/L),准确称取 。0.0100 g 咖啡因标准品于100m L容量瓶中,用水溶解并定容至刻度,作为标准储备液。根据需要再用水将标准储备液稀释成适当浓度的标准工作液。3.仪器和设备3.1高效液相色谱仪配紫外检测器。3.2超声波振荡器。4.测定步骤4.1 提取称 取 0.1g(准确至 0.0001 g )均匀试样于 100m L容量瓶中,加人 80m L水,置超声波振荡器中超声20 min,冷却后用水定容至刻度并混匀,过0.45滤膜后,供HPLC测定。4.2 测定4 .2 .1 色谱条件a) 液 相色谱仪,配紫外检测器,检测波长273n m;b) 色 谱柱:C,。柱(25cm×4.6m mID,5um)柱或相当柱;c) 流 动相:乙睛一水一乙酸(16+83十1);d) 流 速 0.5 m L/min;e) 进 样 量 5uL4.2.2 色谱测定根据样液中咖啡因的含量情况,选定峰面积相近的标准工作溶液。标准工作溶液和样液中的咖啡因的响应值均应在仪器的检测线性范围内对标准工作溶液和样液等体积参插进样测定。在上述色谱条件下,咖啡因的保留时间约为9min。 http://ng1.17img.cn/bbsfiles/images/2013/07/201307291536_454528_2764104_3.jpg 标准溶液色谱图 http://ng1.17img.cn/bbsfiles/images/2013/07/201307291537_454529_2764104_3.jpg 标准曲线 http://ng1.17img.cn/bbsfiles/images/2013/07/201307291537_454530_2764104_3.jpg 样品色谱图 4.2.3[/size
麻醉剂苯佐卡因的合成目的原理实验目的1.学习多步骤有机合成实验线路的选择和实验方法;2.学习掌握回流、过滤等操作技术。实验原理苯佐卡因(Benzocaine)是对氨基苯甲酸乙酯的俗称,可作为局部麻醉药物,以甲苯为原料可以有三种不同的合成路线制的苯佐卡因。第一条合成路线步骤多,产率较低;第二、三条战线则步骤较少,产率高。尤以第二条线路效果最佳,具有实验步骤少、操作方法、产率高的优点,也可利用前面一般合成中的产品(对硝基苯甲酸)作为原料,可节约药品。采用第二条路线,以对硝基苯甲酸为原料,通过先还原后酯化制得苯佐卡因,反应分为两步:第一步是还原反应HOOC-Ar-NO2 +Sn + HCl → HOOC-Ar-NH2HCl + SnCl4以对硝基苯甲酸为原料,锡粉为还原剂,在酸性介质中,苯环上的硝基还原成氨基,产物为对氨基苯甲酸。这是一个既含有羧基又含有氨基的两性化合物。故可通过调节反应液的酸碱性将产物分离出来。还原反应是在酸性介质中进行的,产物对氨基苯甲酸形成盐酸盐而溶于水中还原反应后锡生成四氯化锡也溶于水中,反应完毕加入浓氨水至碱性,四氯化锡沉淀可被滤去SnCl4 + 4NH3H2O → Sn(OH)4 + 4NH4Cl而对氨基苯甲酸在碱性条件下生成羧酸铵盐仍溶于水。然后再用冰乙酸中和过滤,而氨基苯甲酸固体析出。对氨基苯甲酸为两性物质,酸化或碱化时都必须小心控制酸碱用量,否则严重影响产量与质量,有时甚至生成钠盐而得不到产物。第二步是酯化反应COOH-Ar-NH2 + C2H5OH + H2SO4 → C2H5OOC-Ar-NH2由于酯化反应有水生成,且为可逆反应,故使用无水乙醇和过量的硫酸。酯化产物与过量的硫酸形成盐溶于溶液中,反应完毕后加入碳酸钠中和即得苯佐卡因固体。仪器药品对硝基苯甲酸,锡粉,浓盐酸,浓氨水,冰乙酸;对氨基苯甲酸(自制),无水乙醇,浓硫酸,硫酸钠;100ml圆底烧瓶,球形冷凝管,250ml烧杯,布氏漏斗,吸滤瓶,培养皿。过程步骤(1) 还原反应称取4g(0.02mol)对硝基苯甲酸,9g(0.08mol)锡粉加入到100ml圆底烧瓶中,装上回流冷凝管,从冷凝管上口分批加入20ml(0.25moL)浓盐酸,边加边振荡反应瓶,反应立即开始(如有必要可用大火加热至反应发生)。必要时可再微热片刻以保持反应正常进行,反应液中锡粉逐渐减少。当反应接近终点时(约20~30min),反应液呈透明状,稍冷,将反应液倾斜倒入250ml烧杯中,用少量水洗涤留存的锡块固体。反应液冷至室温,慢慢的滴加浓氨水,边滴加边搅拌,使溶液刚成碱性。过滤除去析出的氢氧化锡沉淀,用少许水洗涤沉淀,合并滤液和洗液,注意总体积不要超过55ml。若体积超过55ml,可在水浴上浓缩。向滤液中小心地滴加冰乙酸,有白色晶体析出。再滴加少量冰乙酸,有更多的固体析出,用蓝色石试纸检验到呈酸性为止。在冷浴中冷却,过滤得白色固体,晒干后称重,产量约为2g。(2) 酯化反应将自制的2g(0.5mol)对氨基苯甲酸放入100ml圆底烧瓶中,加入20ml(0.34mol)无水乙醇和2.5(0.045mol)浓硫酸(乙醇和浓硫酸的用量可根据每人得到的对氨基苯甲酸的多少而作相应调整)。将混合物充分摇匀,投入沸石,水浴上加热回流一小时,反应液呈无色透明状。趁热将反应液倒入盛有85ml水的250ml烧杯中。溶液稍冷后,慢慢加入碳酸钠固体粉末,边加边搅拌,使碳酸钠粉末充分熔解,当液面有少许白色沉淀出现时,慢慢加入10%碳酸钠溶液,将溶液pH值调至成中性,过滤得固体产品。用少量水洗涤固体,抽干,晾干后称量。产量1~2g。分析思考 1.如果判断还原反应已经结束?为什么?2.酯化反应为何先用固体碳酸钠中和,再用10%碳酸钠中和反应液?
各位高手可有盐酸罗哌卡因红外谱图啊?还有顺便问下,药典上说药品1-2mg氯化钾200mg,其中[color=#d40a00]1-2mg[/color]和[color=#d40a00]200mg[/color]只是个用质量来描述固体样品量的方式吧,概数,无需用天平来称量、量化吧?正如液体样品可用体积的量来描述,L,ml或滴。是这样理解的吧?
测定药品(丙胺卡因,利多卡因)中二氯甲烷和N,N-二甲基甲酰胺
大家有测过盐酸利多卡因原料含量的吗?(2005版中国药典二部中的方法),在测定含量时颜色的变化明显吗?
想问一下别的实验室,有做过咖啡制品中咖啡因含量的检测吗?我主要是想知道不同类型的咖啡制品中咖啡因含量大致有多少?比如黑咖啡,白咖啡等等。
去过不少城市,也办了不少银行卡,有几个银行卡几年没有用过了,不过听说银行有小额账号管理费,银行卡少于300元,每季度收取3-5的管理费?这怎办呢?
各位前辈,小弟刚接触果醋饮料,请问果醋饮料的检测指标有哪些阿,还请各位不吝赐教阿,谢谢了。
[center]近日英国NICE建议应用阿达木单抗治疗银屑病[/center]近日,英国国家卫生与临床评价机构(NICE)公布一份指南草案:对符合一定条件的严重银屑病成人患者,可以用阿达木单抗 (adalimumab )作为一种选择治疗方法。 该指南草案限制对标准全身治疗无应答或对这类治疗不耐受或有禁忌证的患者使用阿达木单抗。标准治疗包括环孢素(ciclosporin)、甲氨蝶呤(methotrexate)及补骨脂素(psoralen)加紫外线照射。NICE的指南草案建议,第16周仍对治疗无应答的患者应停止阿达木单抗的治疗。 在以往的相关指南中,NICE向同类人群推荐依那西普(etanercept)。在最近的评价中,该机构委员们称,不做出依那西普优于阿达木单抗的推荐建议,医生可根据临床情况在这两种产品中进行选择。 阿达木单抗治疗患者应是适用抗肿瘤坏死因子的治疗人群,此类患者病情严重,NICE定义为银屑病区严重指数(PASI)达到10或超过10,皮肤病生活质量指数(DLQI)超过10; 对阿达木单抗有明显应答的银屑病定义为,治疗期间PASI数值降低75%,或PASI值降低50%且DLQI减少5个点。卫生专业人员采用DLQI数值制定治疗方案时要考虑患者的身体残疾情况或者语言或沟通困难程度。 信息来源:中国医药123网
咖啡因 精密称取上述细粉适量(约相当于咖啡因150mg),置分液漏斗中,加稀硫酸20ml,振摇数分钟使咖啡因溶解,用三氯甲烷提取4次(40ml、30ml、20ml、10ml),合并三氯甲烷液,置于水浴上蒸干,残渣加稀硫酸30ml,回流1小时,放冷后移入100ml量瓶中,用水稀释至刻度,摇匀,过滤。精密量取续滤液40ml置100ml量瓶中,精密加碘滴定液(0.05mol/L)50ml,用水稀释至刻度,摇匀,在约25℃避光放置15分钟,滤过,精密量取续滤液50ml,用硫代硫酸钠滴定液(0.1mol/L)滴定,至近终点时,加淀粉指示液2ml,继续滴定至蓝色消失,并将滴定的结果用空白试液校正即得。每1ml碘滴定液(0.05mol/L)相当于5.305mg的C8H10N4O2·H2O。 ﹙V0-V﹚×C×5.305×100/50 计算公式: W×0.05V0为空白试验时消耗硫代硫酸钠滴定液的体积(ml);V为样品测定时消耗硫代硫酸钠滴定液的体积(ml);C为硫代硫酸钠滴定液的浓度(mol/L);W为样品的称量(g);这个公式是否正确,大家一起讨论啊。。。。。。。。。。。。。。







 我要推广仪器
我要推广仪器
 下载APP
下载APP