怎样进行硼酸酯中硼酸的含量测定本实验室正在进行二乙二醇乙醚和硼酸反应的试验,最终想确定反应剩余的硼酸的量。不知道用什么分析方法定量,希望各位高人指教![em06]
如题 硼酸类是如何衍生化?不衍生化 气相检测有困难硼酸酯 如何检测的?分析过的可以分享一下过程
本人在一家化工厂做实验,最近发现实验偏出理论预期(当然是变坏),初步怀疑可能是硼酸三丁酯和正丁基锂工业品中杂质含量过高或含不利于反映进行的杂质,但鉴于这两种物质的特殊性(氧化性过强),不知该选用何种方法检测或标定,不知道有没有愿意指点一下?由于小弟是新手没几个钱请大侠将就一下啦!奉送五十两纹银!
想配四硼酸钠溶液,因为没配过,上网查的结果是硼砂是含10个结晶水的四硼酸钠,正常情况下含若干个结晶水,在350度下烘3-4小时就成了无水四硼酸钠。按常规配制溶液的方法,就应该用无水四硼酸钠配溶液,硼砂经过3-4小时的烘烤成了爆米花一样的,膨胀很多倍。很轻,想问问有配制经验的版友,是不是就用这个爆米花样的无水四硼酸钠配四硼酸钠溶液?
哪位老师有硼酸三丁酯检测方法,可否提供一下!感谢
四硼酸钠(硼砂)常见都是含结晶水的,配制试剂应该用无水四硼酸钠配制,是用烘烤的方式制备无水四硼酸钠吗?
要测硼酸酯类化合物,带苯环,气化不了,据说遇水硼酸会掉下来。该怎么检测呢
看了一篇文献上说到了偏硼酸锂,如何制作??没有卖的
CZE中需要用到50mM pH9.3的硼酸缓冲液,根据BuffrCal软件给的配制方法,是先用硼酸配成硼酸缓冲液,再加NaOH调至pH9.3。但是周围有人提出硼酸缓冲液应该是用硼砂配制,再调pH,请教一下各位的硼酸缓冲液是如何配制的呢?另外,pH8.0的Tris-硼酸缓冲液又是怎么配制的?我需要50mM的浓度是不是就是1L水中各加50mmol Tris和50mmol硼酸或硼砂?
实验室配制硼酸的作用是什么呢?
求硼酸三丁酯的红外标准谱图!或二价钴氧键的特征吸收峰,谢谢 !!
最近在做皮革中有机锡分析实验,但是遇到一个难题,了解了四乙基硼酸钠的性质和安全说明,由于其在空气中易燃,对空气敏感,对水较敏感,国标是用水来溶解,也有用四氢呋喃溶解配制的,但是由于四乙基硼酸钠特殊的性质,不知道该如何来配制其标准溶液,因为标准品一旦打开了,就暴露在空气中了,如何称量呢? 哪位大侠做过这个实验,教教俺吧,多谢了!说下您配制四乙基硼酸钠的具体过程?
今年新进的新厂家的四硼酸锂,在进行样品采购前试验阶段还没有感觉出问题,主要是用水泥来确定四硼酸锂的质量。正式订货后开始使用后,在分析熟料时感觉出炉的熔样发粘,气泡摇不出去,样品在熔样过程中熔样不匀(尤其是硅熔的不好,表现仪器分析出的氧化硅高、低不均,对率值产生影响),各位有什么好的办法吗?注:1、此次新采购的四硼酸锂细度极细,200μR可达4%;2、粉磨后的熟料,32μR达13%,符合制片要求;3、这次新采购的四硼酸锂所含杂质极少,烧失量也小。
如题。配置0.25M的。标准中称取100.00g,定容至1000mL。四硼酸钠是带10个结晶水的,称量是折算成称取约135g,但是不溶解,加热溶解,一恢复常温就结晶析出,影响含量。怎么办呢
硼酸怎么配置能中和HF?
0.1%甲基红乙醇溶液70ml0.1%溴甲酚绿溶液100ml1%硼酸溶液10L配置的吸收液,有悬浮物,应如何配制才能让该吸收液没有悬浮物?
有哪位大侠做过三(三甲基硅烷)硼酸酯或三(三甲基硅烷)磷酸酯的分析啊?用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的话选用何种柱子合适啊?
[color=#444444]最近在做一个项目,其中一个起始物料的结构带有硼酸酯,试过不同pH值的缓冲液,液相走出来均是不成形的峰,不知道有没有大神做过类似结构的东西,能否分享一下色谱条件借鉴一下,谢谢![/color]
最近在做皮革中有机锡分析实验,但是遇到一难题,了解了四乙基硼酸钠的性质和安全说明,由于其在空气中易燃,对空气敏感,对水较敏感,国标是用水来溶解,也有用四氢呋喃溶解配制的,但是由于四乙基硼酸钠特殊的性质,不知道该如何来配制其标准溶液,标准品一旦打开了,就暴露在空气中了,怎么称量它呀?哪位大侠做过这个实验,教教俺吧,多谢了!
请问大家测试硼酸里的杂质直接可以用超纯水溶吧,第一次测这个不知对仪器是否有污染?
1.容易掉粉末,污染密封圈、铍窗、顶杆plunger凹槽、转盘turret。2.由于压片操作规范程度不易控制,造成测量面有缺陷: a、被硼酸粉末污染; b、测量面不完整;3.由于垫片加工精度不够,测量面边沿有凸出薄壁使压片放不平。4.长期使用硼酸对分光晶体有腐蚀。5.还是用钢环好。
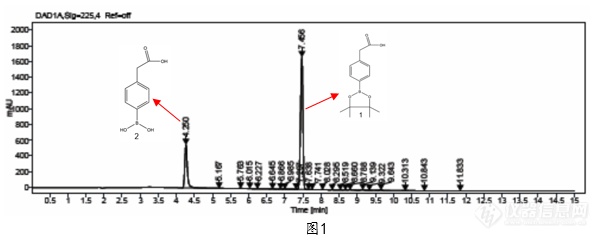
[align=left]近期我们遇到了一种硼酸酯类的化合物1,采用实验室通用方法进行检测的时候发现会出现一个很大的杂质2,根据工艺分析不可能会出现这么大的杂质,定量核磁检测发现该物质含量比较高,并不存在这个大的杂质,用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]去鉴定后发现该杂质为该化合物的水解杂质2(如图1)[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090922409286_9676_5310417_3.png[/img][/align][align=center]图 1:流动相A: 0.05%TFA 流动相B: ACN条件下的样品色谱图[/align]为此我们判定肯定是检测方法出现了问题,首先我们排除稀释剂的影响,稀释剂为乙腈,做了相应的稳定性实验,发现临用新配情况下该杂质仍旧很大。由此我们判断可能是流动相导致该化合物1不稳定会水解生成杂质2。考虑到硼酸酯类化合物可能对酸不稳定,在酸性条件下会被催化水解成硼酸类化合物和相应的醇,因此打算更换其他流动相。首先我们尝试了碱性体系(如图2),由于该化合物1为酸性化合物,在碱性条件下保留较弱,但是从图谱可以看出水解杂质仍旧比较大,由此可以判断在碱性条件下该化合物1也并不稳定。[align=left][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090922406680_5272_5310417_3.png[/img][/align][align=center]图 2:流动相A: 0.1%NH4OH 流动相B: ACN条件下的样品色谱图[/align][align=left]随后我们又尝试了中性体系,采用中性体系的流动相进行测试(如图3)。从图3(a)可以看出,水做流动相条件下,由于流动相的离子强度不够导致峰形丑,还可以看出水解杂质2仍旧存在,但从(b)中可以看出当用乙酸铵作为流动相时候,峰形对称,水解杂质2也比较小。[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090922412671_1242_5310417_3.png[/img][/align][align=left][/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090922413707_3568_5310417_3.png[/img][/align][align=center]图 3:(a)流动相A: 水 流动相B: 乙腈 (b)流动相A: 10mM 乙酸铵水溶液 流动相B: 乙腈条件下的样品色谱图[/align][align=left]根据以上结果我们猜测:该化合物对酸碱都不稳定,但中性条件下只在乙酸铵体系下稳定,为此我们从化合物1本身及水解杂质2的结构分析,该化合物1中的硼原子为sp2杂化,还存在一个空的p轨道,这个空轨道易于接受水和醇等带有未共用电子对的亲核试剂的进攻而使硼酸酯水解([font='adobeheitistd-regular'][size=13px]其机理见方程式[/size][/font][font='dlf-32769-4-2073904376+zipdfa-8'][size=13px]([/size][/font][font='dlf-3-0-25052658+zipdfa-84']1[/font][font='dlf-32769-4-2073904376+zipdfa-8'][size=13px]))。[/size][/font]继续与水作用,生成相应的醇和硼酸。[/align][align=left][/align][img]" style="max-width: 100% max-height: 100% [/img]通过对此分析,似乎已经能够解释化合物1对碱不稳定的原因,即羟基中氧上的孤对电子会进攻硼的空轨道导致其水解,至于为什么在乙酸铵体系中是稳定的我们推测原因是乙酸铵的氮原子会与硼原子形成配对键,从而使该化合物1稳定。 虽然只是硼酸酯类化合物中的一种物质的检测,但是根据检测结果和分析可以为以后的该类化合物的方法开发提供思路,即通在对硼酸酯类的化合物进行方法开发时候,尽量不要采用酸碱体系的流动相,可以考虑用乙酸铵缓冲液作为流动相进行检测。[align=left] [/align]
高硼硅酸盐玻璃、电子、精细硼化物、含硼新材料等行业对硼酸的需求量几乎占国内需求总量的65%以上。但是,这些行业几乎全部使用俄罗斯、美国、土耳其和智利四国的产品,对国产硼酸则不予采用,以致使我国硼酸市场60%的份额被进口产品所控制。 为什么会出现上述情况呢?究其原因有以下几点: 1、质量与进口产品有明显的差距。 近几年来,我国以硝酸为分解剂、以硼砂为原料的两步法硼酸发展较快,有少数企业的产品质量勉强能够满足上述行业的要求。以硫酸为分解剂、以硼镁矿为原料的一步法硼酸,产品质量差距较大,难以为上述行业所接受。辽—吉地区的生产企业为了提高收率,近年来采用了浮选法工艺,该工艺虽然明显的提高了硼的收率,但却给硼酸的产品品质带来了不利影响,一是产品粒径较小,外观质量较差;二是含铁超标;三是硫酸盐超标,根本就不能在上述领域应用。应该说,和前几年相比,该地区的硼酸生产收率提高了,品质却下降了。 青海地区硼酸生产发展较快,但由于科技基础薄弱,工业基础差,加之科技投入不足,生产企业普遍存在着技术含量低、设备简陋、生产水平不高、产品质量差等问题。所产产品色泽灰暗,水不溶物、铁、硫酸盐含量超标,只能应用于玻球、玻纤、冶金和农业领域。该地区有际华江源公司、辰光公司等企业比较注意科技投入和技术进步,产品质量达到智利同类产品标准。 我国硼酸产品目前执行的是GB538-2006标准,GB538-1990标准早已于2007年2月1日废止。但大部分生产企业仍然按90标准进行生产。从现实情况看,我国硼酸品质和进口产品相比,差距有越来越大的趋势。 2、质量不稳定。 据有关用户介绍,国产两步法硼酸的质量是能够达到或接近进口产品标准的,但问题是产品质量缺乏稳定性。一些企业虽然能够生产出符合高端领域应用的产品,但由于受工艺控制、生产管理等因素的影响,产品质量波动较大,这给用户带来了使用上的困难。因此,用户为了稳定自身的工艺条件、保障自己的产品质量而不愿使用国产产品。
用于络合氢氟酸所用的饱和硼酸怎样配制?室温下称量多少质量的固体硼酸溶解于去离子水中?谢谢
0.1%甲基红乙醇溶液70ml0.1%溴甲酚绿溶液100ml1%硼酸溶液10L配置的吸收液,有悬浮物,应如何处理?应如何配制才能让该吸收液没有悬浮物?顺序是怎样呢
最近检测3-吡啶硼酸片呐酯,用C18柱,254nm,乙腈比水9比1流动相,检测时,峰高只有60,而且拖峰严重,请问一下这个物质最大吸收波长大概是多少,用什么柱子检测影响大不大。物质的结构在附件里,请大家帮一下啦。
[b][font=宋体]问题描述:一个起始物料的结构带有硼酸酯,试过不同[/font]pH[font=宋体]值的缓冲液,液相走出来均是不成形的峰,是什么原因?有没有合适的色谱条件?[/font][font=宋体]解答[/font]:[/b][font=宋体]([/font]1[font=宋体])先了解硼酸酯在液相中为什么走不出来,因为硼酸酯化合物遇水和醇后都极易水解,而我们常规反相色谱使用的流动相为水(或酸[/font]/[font=宋体]碱[/font]/[font=宋体]盐溶液)[/font]+[font=宋体]有机相,所以在检测硼酸酯样品的时候很容易出现峰不成形。[/font][font=宋体]([/font]2[font=宋体])根据硼酸酯的特性我们得知,液相正相色谱可以很好的检测,并且根据查询文献可知,已经有研究表明硼酸酯类化合物可以采用非水的反相检测方法。其实只要采用有机溶剂检测硼酸酯类化合物的出峰时间不是太靠前,完全可以采用反相色谱配普通[/font]C[sub]18[/sub][font=宋体]色谱柱进行检测。[/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font][color=red] [/color]
0.05M的硼酸硼砂缓冲溶液ph在5至10之间能配吗?怎么配?希望大家帮忙解决一下 !
请教一下大家:Tris-硼酸缓冲液是怎么配制的?文献中写的是Tris-borate pH8.5,怎么配制?
请问,如要测试硼酸中的铁和铅,在前处理方面是直接加水和酸溶解还是要进行特别一点的操作?谢谢!







 我要推广仪器
我要推广仪器
 下载APP
下载APP