食品中氨基酸测定的国标中,对样品脂肪含量都进行了要求。即样品的脂肪含量必须低于4-5%才能进行水解,否则要先脱脂。想请教一下脱脂的方法是什么,只称50mg的乳粉,也需要脱脂吗?在各种文献上找,都说需要脱脂,并没有看到具体的做法那,打扰各位专家了!非常感谢!
视频中氨基酸测定的国标中,对样品脂肪含量都进行了要求。即样品的脂肪含量必须低于4-5%才能进行水解,否则要先脱脂。想请教一下脱脂的方法是什么,只称50mg的乳粉,也需要脱脂吗?在各种文献上找,都说需要脱脂,并没有看到具体的做法那,打扰各位专家了!非常感谢!
我用氨基柱在做反相色谱时,氨基柱的出峰顺序和C18 的出峰顺序相反,请问氨基柱色谱拖尾时,该怎么处理?我的流动相是乙腈:柠檬酸缓冲盐(1.35g柠檬酸至1000ml水,三乙胺调节PH至5.0)=77:23
氨基柱通常都是用不低于50%的有机相冲洗柱子,这样能否保证杂质洗脱的干净呢?
请教各位专家。氨基酸测定的国标中,对样品脂肪含量都进行了要求。即样品的脂肪含量必须低于4-5%才能进行水解,否则要先脱脂。想请教一下脱脂的目的是什么,脂肪的存在对氨基酸的测定有哪些不利影响?谢谢。
现有基因重组表达的糖蛋白想进行以下几项委托检测, C端氨基酸测序、氨基酸组分分析、肽图、质谱分子量、糖基化分析如果有意者请联系,将样品要求、检测费用以及合作流程发到邮箱,如有疑问可以加qq联系。 qq:278569901 邮箱:wbz5102@gmail.com
[b]【求助】季铵碱的HPLC氨基柱分离拖尾严重如何解决[/b] 季铵碱的HPLC氨基柱分离拖尾严重如何解决做了一个周的甜菜碱的HPLC方法,用了NaH2PO4(PH=4.5)的C18反相柱分析,又用了S5-NH2柱的正相乙腈:水(85:15)做对照,均出现对照拖尾严重的问题,加了0.1%TFA仍没改善,请问各位大虾,如何解决????急求?氨基柱可以加三乙胺防拖尾吗???谢谢!
新买了一个氨基柱(Hypersil GOLD Amino),用来分离单糖类化合物,在液质联用仪器上使用。当使用乙腈:水=80:20的洗脱溶剂进行洗脱时,发现出现两个很高的质谱信号138.0573以及120.0471,前者感觉是氨基苯甲酸,后者不知道是什么。138的信号特别强,达到了8E9。当使用纯的乙腈溶剂洗脱时则上述两个信号消失,不知道是不是洗脱溶剂中有水时造成氨基柱水解的缘故,不知道该怎么处理,希望高手帮忙
有朋友做过2-丙烯酰氨基-2-甲基丙烷磺酸的液相分析吗?请指点,谢谢!
求助6-氨基青霉素烷酸中蛋白质检测 ,也就是6-APA中蛋白质含量检测,有做过的给说下吧,谢谢
[color=#444444]样品是:3-氨基-2-恶唑烷酮水溶液,现有分析方法是HP-5色谱柱,自动进样1.0ul,主峰峰太宽,目前正在优化方法,有做过的给些意见,万分感谢[/color]
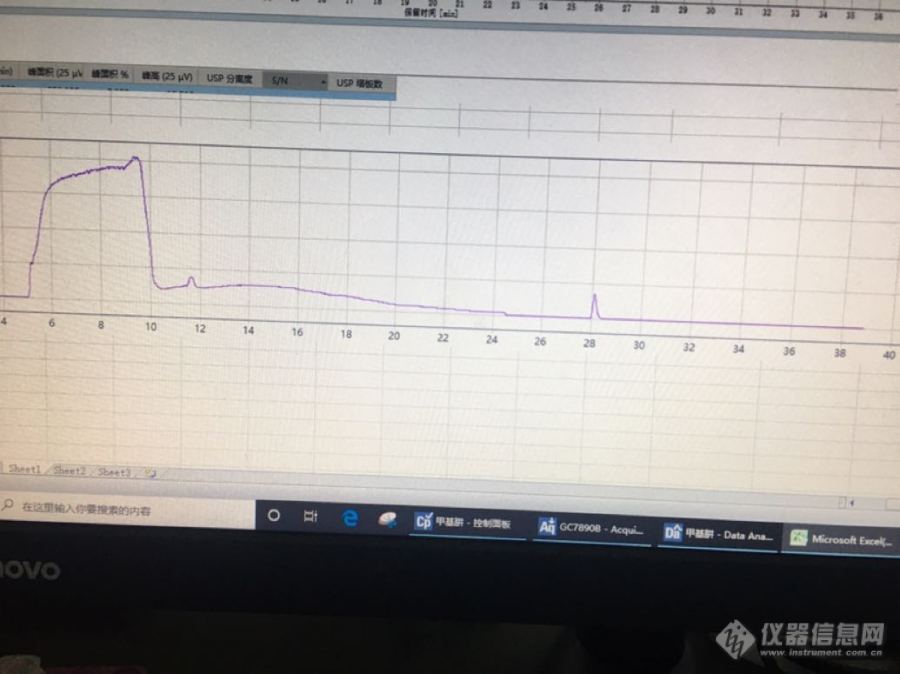
新手,最近在做中控方法开发,原料为金刚烷胺盐酸盐,产品为3氨基金刚烷醇,查到了原料的药典标准,进样口220,程序升温70,5min,10度的速率升到250,保持17min,总共40min,用的TCD检测器,药典里用的FID,因为还有别的项目在做所以一起用的TCD,温度300。柱子用的SE30,原料能出峰,但是产品会在运行结束之后过一段时间才出峰,请问大家怎么调方法,能让这俩物质一起出峰。。。金刚烷醇熔点大概266左右[img=,690,516]https://ng1.17img.cn/bbsfiles/images/2022/03/202203301000579912_59_3989875_3.png[/img]
文献中使用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]来检测ω-氨基十一烷酸,需要用到含棕榈酸的乙酸乙酯,但是买回来的ω-氨基十一烷酸不溶于乙酸乙酯怎么办?
[color=#444444]自动氨基酸分析仪测素丸子里的氨基酸的样品前处理的方法 或者同类也行[/color]
做农残灭幼脲,前处理要过氨基萃取柱,别的实验室用二氯甲烷和甲醇95:5可以很好的洗脱回收,我用这个比例用我的萃取柱就什么都洗不下来,上机没有灭幼脲,他们用的迪马的萃取柱,我们是安捷伦的萃取柱,怎么就不行呢?什么原因?
我要测定植物激素,文献上说需要使用聚乙烯聚吡咯烷酮(PVPP)柱、二乙基氨基乙基交联葡聚糖凝胶柱。我没有用过,请问这种柱子一般怎么用啊?上样量多少,用什么洗脱?怎么保存?怎么预处理?谢谢啊。文献上都没有写。谢谢啦。文献我上传了,请大家帮帮我啊。非常感谢。 [~188310~]
各位大侠,小弟刚接触液相,有个问题想请教下,带我做实验的测样前和测完样都是用10%的乙腈冲氨基柱,流动相是乙腈-0.05 M KH2PO4 60:40,请问这样冲对氨基柱伤害大吗?总觉得这样不妥。
我的实验需要从氨基酸底物中鉴定N-乙酰氨基酸的生成和生成的量,有那位高手知道用什么柱子和那一种洗脱液处理吗?是否有更简单的办法?比如类似茚三酮的显色方法吗?万分感谢你的回答.谢谢
用氨基柱做糖类化合物,加什么可以减轻拖尾,流动相是:乙氰:磷酸盐缓冲液。现在的情况是拖尾还是严重啊。怎么办呢?还有就是如何快速平衡色谱柱呢?
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]做GB5009.97-2016食品中环己基氨基磺酸钠的测定,有人说把正庚烷换成正己烷比较好,他们说正庚烷不如正己烷纯,很容易出现杂峰大家怎么看
(2S,3S)-3-氨基-2-甲基-4-氧代氮杂环丁烷-1-磺酸(2S,3S)-3-amino-2-methyl-4-oxoazetidine-1-sulfonic acid谁有这个液相的检测纯度的方法,是氨曲南的起始物料N-氮杂环丁烷???
农残检测,比如说吡虫啉、多菌灵和氨基甲酸酯类农药检测,样品处理应该选择哪种柱子进行纯化,假如用氨基柱纯化,洗脱液怎样选择梯度?我最后一般用的都是甲醇+二氯甲烷(5+95),这些农药在水里和有机溶剂里都能部分溶解和全部溶解,我就没有进行梯度洗脱,以致样品在检测时杂峰很多
新买了一个氨基柱(Hypersil GOLD Amino),用来分离单糖类化合物,在液质联用仪器上使用。当使用乙腈:水=80:20的洗脱溶剂进行洗脱时,发现出现两个很高的质谱信号138.0573以及120.0471,前者感觉是氨基苯甲酸,后者不知道是什么。138的信号特别强,达到了8E9。当使用纯的乙腈溶剂洗脱时则上述两个信号消失,不知道是不是洗脱溶剂中有水时造成氨基柱水解的缘故,不知道该怎么处理,希望高手帮忙
流动相选择:流动相A为醋酸盐-磷酸盐缓冲溶液(pH=4.95);流动相B为乙腈;流动相C为超纯水。检测氨基酸混标(含17种氨基酸)时,出现34个峰,猜测梯度洗脱程序设置不合理。
使用ACCQ.TAG 氨基柱检测甘氨酸含量,内标物质α-氨基丁酸拖尾因子比较大(药典标准0.95-1.40),新柱子开始做拖尾因子就1.3多,做三四次后拖尾因子就超标准了,请教各位大神是什么原因?
请问用万通785滴定仪测氨基酸态氮怎么设置参数?就是pH=8.2,pH=9.2。但是中间加甲醛的时间怎么控制?可不可以设置我加完甲醛,再开始滴到9.2呢?
[b]【求助】季铵碱的HPLC氨基柱分离拖尾严重如何解决[/b] 季铵碱的HPLC氨基柱分离拖尾严重如何解决做了一个周的甜菜碱的HPLC方法,用了NaH2PO4(PH=4.5)的C18反相柱分析,又用了S5-NH2柱的正相乙腈:水(85:15)做对照,均出现对照拖尾严重的问题,加了0.1%TFA仍没改善,请问各位大虾,如何解决????急求?氨基柱可以加三乙胺防拖尾吗???谢谢!
最近在做氨基酸的提取,得到的溶液中有混合氨基酸、寡糖、寡肽等小分子极性成分,现在想纯化氨基酸,不知道该怎么办,求助各位高手,谢谢! PS:以后打算上工艺的,希望大虾们提供些工业上能用的,多谢!拜托!
问题: 有人测脱氢乙酸吗? 有人测过这些吗?如图, 我想了解哪个水相高点? 氨基甲酸酯不太了解
液相测维生素C,用氨基柱,流动相是甲醇与柠檬酸,但是峰拖尾严重,怎么改善?







 我要推广仪器
我要推广仪器
 下载APP
下载APP