http://simg.instrument.com.cn/bbs/images/default/em09511.gif如题,求乙酰唑胺的EP5.0。
我是新手,做乙酰甲胺磷时,1ppm不出峰,增加浓度,峰很宽。不知该如何继续下去?
用NY761方法做乙酰甲胺磷与甲胺磷的回收率,标液用丙酮配制,衬管用高惰性衬管,不是新的,是用过的,添加量为0.16ug/mL,甲胺磷实际测得为0.20ug/mL,乙酰甲胺磷为0.35ug/mL,回收率为125-216%,为何回收率这么高,是因为高惰性衬管受到污染了吗?还是必须用基质配标液。
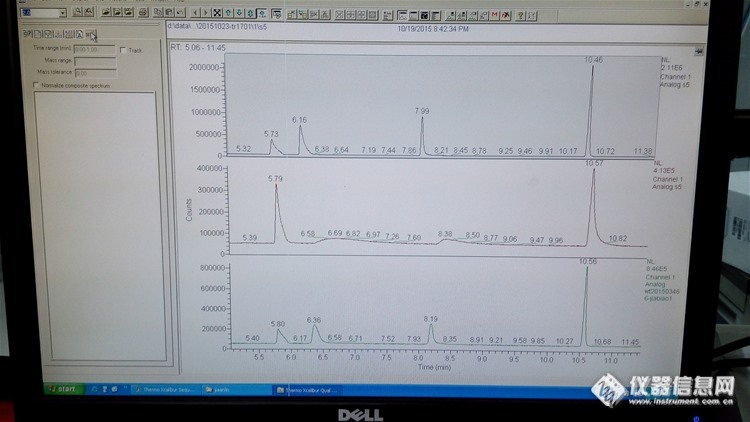
http://ng1.17img.cn/bbsfiles/images/2015/11/201511111107_573060_2547863_3.jpg如图,第一个谱图是1个月前做的标准曲线点,峰分别是敌敌畏,甲胺磷,乙酰甲胺磷,乐果,用的柱子是35柱第二个谱图是前天做的标准曲线点,其中甲胺磷,乙酰甲胺磷峰塌下去了第三个谱图是前天做的红枣加标样品,其中甲胺磷,乙酰甲胺磷峰很正常。如今往红枣样品上机液(四个峰均不出峰)添加混标液,得出的谱图和第三个一样;重新配了混标,甲胺磷和乙酰甲胺磷也是超级拖尾,但是一旦把混标加入样品液里面,两个出峰都很正常求解决方法(已经切割过柱子前端,进样瓶都是新的,除了溶液一个是丙酮定容的样品上机液,一个是色谱纯丙酮不一样外,其他条件都一样)
求乙酰肼,恶二唑酮,唑丙酮,三嗪酰胺检测方法,跪求,谢谢
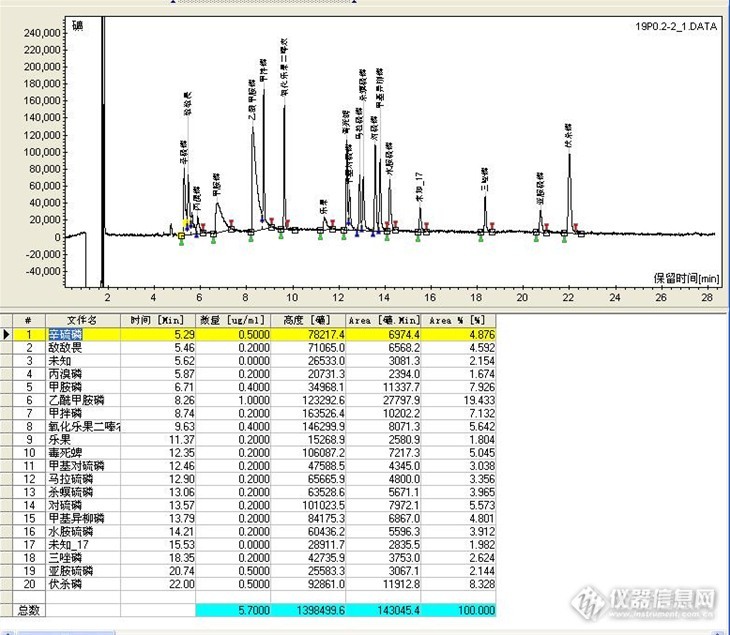
用FPD检测器,DB1701色谱柱做乙酰甲胺磷时,容易被衬管吸附,用高惰性衬管出峰,但峰有拖尾现象,那么有没有人用别的型号的色谱柱做乙酰甲胺磷,检出限低而出峰尖锐?http://ng1.17img.cn/bbsfiles/images/2013/05/201305231603_441225_1645480_3.jpg这是DB1701(30*0.25*0.25)色谱柱的乙酰甲胺磷出峰情况。
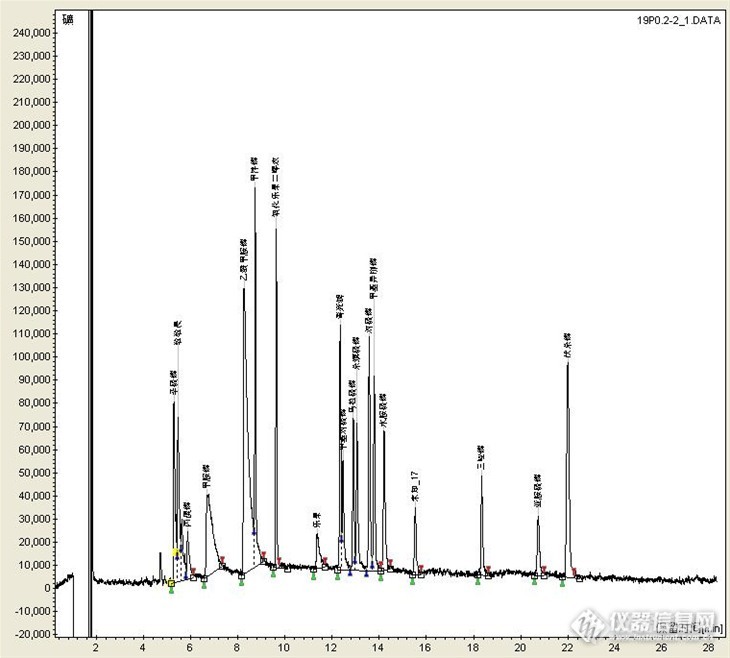
用FPD检测器,DB1701色谱柱做乙酰甲胺磷时,容易被衬管吸附,用高惰性衬管出峰,但峰有拖尾现象,那么有没有人用别的型号的色谱柱做乙酰甲胺磷,检出限低而出峰尖锐?http://ng1.17img.cn/bbsfiles/images/2013/05/201305231601_441223_1645480_3.jpg这是DB1701(30*0.25*0.25)色谱柱的乙酰甲胺磷出峰情况。
最近做乙酰甲胺磷发现一个奇特的现象。方法如下,乙腈提取提取,浓缩后过某大品牌氨基柱,乙腈甲苯3:1淋洗,浓缩后甲醇水定容,上LCMSMS,定量用基质标。基质为普通白菜。上机试液理论值100ppb,结果乙酰甲胺磷没有出来,开始以为氨基柱的问题,后来一个突发想法,把试样稀释了十倍,再上机分析,乙酰甲胺磷回来了,而且值为6~8ppb,符合回收率。LCMSMS为watersuplc,TQD,C18液相柱。请问各位同行,有用液质做乙酰甲胺磷的大侠吗?基质效应那么大?
最近做绿茶提取物甲胺磷,乙酰甲胺磷,对硫磷的检测,回收率做不出,甲胺磷乙酰甲胺磷才4-5%,对硫磷有70-80%。我使用的是安捷伦的6890N,检测器NPD,不分流进样,进标准品1.0ug/ml峰面积大概是300左右,但是不是很稳定。感觉问题出在前处理,分别用正己烷,乙酸乙酯,乙腈做过过加样回收,加2ml浓度1.0ug/ml。乙酸乙酯,乙腈提取浓缩后为粘稠红棕色液体,用无水硫酸镁和活性炭分散固相萃取后没有改善,正己烷提取后浓缩液无色。浓缩前进过仪器是可以测出来的,浓缩后没有了,回收液液没有,估计分解了。浓缩使用的是步其的平行定量浓缩仪,水浴45度,真空度250Mbar,冷凝水10-13度。我认为是浓缩步骤出的问题,大家帮分析一下,到底是哪里出的问题?有没有跟好的前处理分享一下,要方法简单的哦
邻氯乙酰乙酰苯胺的国标或行标
我用的是安捷伦[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]7890A,做乙酰甲胺磷时为什么做不出来,需要在什么样的条件下检测?
本人刚做[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]没多久,碰上单位新买一台安捷伦7890A的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],今天第一次用它来做杀螟硫磷和乙酰甲胺磷,色谱条件为氢气3ml/min,空气60ml/min,尾吹7ml/min,检测器为NPD温度为325,程序升温:150保持2分钟以8℃/min升到250再保持12分钟,使用液浓度为0.1ug/ml(混标),柱子是DB-17,但做出来很不好,杀螟硫磷的响应值只有约10PA, 乙酰甲胺磷更小,问题出在哪里吗?有劳各位高手帮分析一下,谢了。
最近做农残,发现0.5的农残混标中甲胺磷乙酰甲胺磷氧乐果峰小的可怜,我也按照论坛上的方法试了,截取了20厘米的柱子,换了新衬管和进样垫,重新配了1ug/ml的标样,还是那样,这几种物质峰特别小,请大家帮忙分析下原因,急!!!
灭蝇胺和乙酰甲胺磷在[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]上的回收率只有百分之四左右,主要是什么原因啊?我直接用水做基质加标,回收率也是啊[em0815]
请问如何提高FPD的响应值?我做的是乙酰甲胺磷的农残检验,我现在的最低响应值为1ppm,再低就没有响应了,但国标中说乙酰甲胺磷的最大残留量为0.2ppm,我根本达不到,求指点
各位大侠,小弟欲请教下有没有用瓦里安2200离子阱气质检测甲胺磷和乙酰甲胺磷,急求方法,我这两天一直在做但是全扫效果都很差,欢迎赐教!谢谢
甲胺磷和乙酰甲胺磷的检出限怎么去换算成含量?GB/T 5009.103-2003[img]https://ng1.17img.cn/bbsfiles/images/2022/07/202207251127261869_1391_3848419_3.png[/img]
结球甘蓝,乙酰甲胺磷,我都不知道该不该判了。。。。。。。。双柱确认就是那个峰啊。。。。。。但是上级单位检测就是没有不知道该怎么办???????以后怎么做呢?????
蔬菜中有机磷农药的检测,新换的衬管分别用用甲胺磷和乙酰甲胺磷单标饱和,然后接着做混标,发现甲胺磷和乙酰甲胺磷的单标和混标中的出峰时间不一致,相差挺大的,单标是3个月前配的(冷藏),以前也没出现过这种情况,请教各问大侠了,谢谢!
不知哪位大哥有乙酰羟胺的分析方法?多谢!
乙酰甲胺磷的问题我直接进样一定系列浓度的乙酰甲胺磷,线性很差,只有0.992,但是进样其他的,譬如敌敌畏,其他的线性都很好,不知道为什么
蔬菜中有机磷农药的检测,新换的衬管分别用用甲胺磷和乙酰甲胺磷单标饱和,然后接着做混标,发现甲胺磷和乙酰甲胺磷的单标和混标中的出峰时间不一致,相差挺大的,单标是3个月前配的(冷藏),以前也没出现过这种情况,请教各问大侠了,谢谢!
甲胺磷、乙酰甲胺磷的FPD信号很小怎么回事?如何解决?
请问大家知道100ppm的甲胺磷、乙酰甲胺磷、氧化乐果的保存期限是多久吗?最近做农残标准曲线,使用的是去年8月份配的100ppm的标样,这三种农残0.1ppm的浓度基本没峰,1ppm的峰面积也明显较去年的小很多,不知是不是分解了。
RT:甲胺磷已经禁用很久了,还是有检出甲胺磷。乙酰甲胺磷中的甲胺磷一般要求在0.8-1%,实际情况是什么样的?
RT:10版药典对乙酰氨基酚中的对氨基酚及有关物质、对氯苯乙酰胺检测有个朋友公司开始做这个项目,药典采用的是C8色谱柱朋友想买热电的柱子,不知道有没有同行采用热电的柱子做过?有的话帮忙发一张图谱看看,谢谢!QQ:342832185
我的是Agilent 7809A,配FPD檢測器,使用DB-1701,30mx0.53mmx0.25um的色譜柱,進樣濃度分別是500ng/ml的甲胺磷及乙酰甲胺磷,響應值分別是8000及5000,而且峰型很好,但是當我轉用HP-5,30mx0.32mmx0.25um色譜柱時,進同樣的濃度,所有條件不變,只是把柱流量調低至1ml/min,甲胺磷及乙酰甲胺磷只是出現微微的駝峰,安全不成峰,同時發現粗直徑較幼直徑的色譜柱適合檢測甲胺磷及乙酰甲胺磷,大家是否有同感,還是我有地方混淆了,大家討論一下
乙酰甲胺磷峰形不好,如何改善?
GC7890A DB1701新柱以及用了一年后截去1米半之后的柱子,FPD检测器,检测器已清洗 进样口已清洗 衬管全新,分流平板全新。5PPM的乙酰甲胺磷峰高1500左右,0.4PPM的峰高180,再低不出峰了。是1701这根柱子对于乙酰甲胺磷吸附很严重,或者说这根柱子不适合分析此种农药。希望大家交流下,说说你们选择分析有机磷农残的毛细柱和色谱条件!
[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]上用玻璃棉少的高惰性衬管,进针次数多以后,乙酰甲胺磷出峰越来越小,换了一种里面没有玻璃棉的衬管,前两针出峰很好,很快也变的很小,乙酰甲胺磷用哪种衬管好?推荐一个高惰性衬管的型号。







 我要推广仪器
我要推广仪器
 下载APP
下载APP