苦瓜胶囊中吡啶甲酸铬的含量测定 吡啶甲酸铬是一种被卫生部允许添加到保健食品中的功效成分之一,具有降糖调脂作用,但加入的量要很好地控制,其中添加过程中的混合均匀度是控制的环节之一,本控制方法采用HPLC法检验其混合混匀度,保证其质量.材料与方法1.仪器Waters 2487检测器-515泵高效液相色谱系统,KWT-100A科伟达超声波发生器, TGL-16G-A离心机, Easypure RF超纯水机.2.试剂磷酸氢二钠(天津大茂化学试剂厂),磷酸二氢钾(天津大茂化学试剂厂),乙腈(MERCK),超纯水3.测试样品苦瓜提取物 : 吡啶甲酸铬(499.9 : 0.1)混合物4.液相色谱条件色谱柱:Agilent Technologies DIKMA ZORBA×SB-Aq 4.6×150mm (LC-002) 柱温:室温 检测波长:254nm 流动相: *0.125mol/L磷酸盐缓冲液:乙腈=9:1 流速:0.5mL/min 进样量:10μL5.吡啶甲酸铬对照品溶液 取吡啶甲酸铬10. 6mg,精密称定,加入甲醇:水(1:1)适量,超声使溶解,并定容至100 mg,作为储备液.6.标准曲线制备 使用以上对照品储备液配制浓度为2.12µ g/ml,5.30 µ g/ml,10.6 µ g/ml,53.0 µ g/ml, 106.0 µ g/ml的系列对照品溶液,在给定的仪器条件下进行色谱分析,以峰面积对浓度作标准曲线. 7.样品的制备: 取苦瓜胶囊粉末1.0g,精密称定,置于具塞锥形瓶中,精密加入甲醇:水(1:1)20ml,称定重量,超声提取5min后,冷却至室温,称定重量,补足失去重量,摇匀,转移至合适的离心管中,3000r/min离心3min,倾出上清液,摇匀,以0.45μm滤膜过滤液即得.
有哪位大神按国标做过保健食品中的吡啶甲酸铬啊~~~~~ 样品处理是甲醇:水 1:1 的溶剂,样品提取很简单就是超声,离心 过滤进样,,,吡啶甲酸铬微溶于水不容易乙醇,,,杂峰很多~~~如何处理
有朋友做过5-氨基-2-吡啶甲酸LC分析吗?请指教,谢谢!
点击链接查看更多:[url]https://www.woyaoce.cn/service/info-19052.html[/url]吡啶甲酸铬检测方法:保健食品中吡啶甲酸铬含量的测定GB/T 5009.195-2003卫生部《保健食品检验与评价技术规范》 2003年版,五、保健食品中吡啶甲酸铬的测定肌醇检测方法:保健食品中肌醇的测定GB/T 5009.196-2003卫生部《保健食品检验与评价技术规范》 2003年版,七、保健食品中肌醇的测定检测周期:5个工作日我中心提供食品安全检测业务范围:食品、保健食品、食品添加剂、特殊医学配方食品、农产品、生活饮用水、食品接触材料、饲料、饲料添加剂等产品。检测项目包括:理化指标、营养成分指标、功效成分指标、微生物指标、农兽药残留、生物毒素、有毒有害物质、非法添加成分,以及食用农产品快检、方法学验证、稳定性试验、实验动物试验(毒理学评价和功效评估)。
最近在做吡啶甲酸铬,国标上的流动相是0.125mol/L的磷酸盐缓冲液,国标上写的是用磷酸二氢钾和磷酸氢二钾配制,没有说怎么配,有人用国标检测过吡啶甲酸铬吗?
3-甲基-2-吡啶甲酸用液相检测时试用了很多种缓冲盐,C18的柱子,峰形一直不好,请教各位有没有做过这个样品或相似的样品,样品具有两性,是不是与氨基酸有点相似?我没有做过氨基酸的样品,请各位给点建议吧。。。先谢谢了^_^
[color=#444444]如题,色谱条件:[/color][color=#444444] 色谱柱:C18 250*4.6*5[/color][color=#444444] 流动相 :磷酸缓冲液(ph3.0、6.0):甲醇=80:20[/color][color=#444444] 波长265nm,[/color][color=#444444]有人做过吡啶甲酸吗,谢谢[/color]

药物名称:吡啶甲酸铬。货号:99603.今日抽奖结果:[align=center][img=,690,295]https://ng1.17img.cn/bbsfiles/images/2019/11/201911151727105147_6492_708_3.png!w690x295.jpg[/img][/align][align=center][img=,690,315]https://ng1.17img.cn/bbsfiles/images/2019/11/201911151727123994_2929_708_3.png!w690x315.jpg[/img][/align][align=center][img=,690,325]https://ng1.17img.cn/bbsfiles/images/2019/11/201911151727136447_8021_708_3.png!w690x325.jpg[/img][/align][align=center]=================================[color=#ff0000]活动规格[/color]====================================[/align][align=left][color=#ff0000]【活动时间】:每个工作日10:00-15:00【活动内容】:根据迪马产品资料:《药物检测应用文集》,每日会出一个化药或中药名称标题,版友根据标题找出相应迪马产品,将从回答正确者中利用抽奖软件抽取以下奖项。[/color][/align][align=left][color=#ff0000]【活动奖励】:一等奖:3个钻石币(2人),二等奖:2个钻石币(3人),三等奖:1个钻石币(5个人)。[/color][/align][align=left][color=#ff0000]【注意事项】:一定要在迪马产品资料《药物检测应用文集》中找出相应迪马产品。[/color][/align]
有谁知道哪个单位有99.9%含量的吡啶甲铬标准品出售?必须附带证书。急需!
在合成对苯二甲酸时加入吡啶做催化剂。现要测定对苯二甲酸产物中吡啶的含量。问:用什么方法?
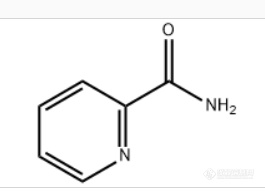
2-吡啶甲酰胺[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]检查,出峰不稳定。求助,现有设备条件能不能准确定量分析?,如果不能,有其他方法吗?感谢大佬解惑!结构式:[img=,265,191]https://ng1.17img.cn/bbsfiles/images/2022/04/202204231127274245_4216_3368413_3.png!w265x191.jpg[/img]色谱条件:进样室:250度;检测器270度。柱温:30度恒温3分,40度升温速率到260度恒温。色谱柱:SE-54(使用温度-20-300)溶剂:异丙醇,内标物:正丙醇。目前只打标液加了2-氰基吡啶(反应物还有2-氰基吡啶,吡啶甲酸甲酯,吡啶-2-亚胺甲酯)问题:峰形不好,出峰不稳定。浓度高峰形前伸,考虑过降浓度,依然峰形没有太好,现在峰面积只有20w,不好再降了。2-吡啶甲酰胺沸点高,250加减30,出峰不稳定,出现下图大宽峰的次数较多,1/3吧求助,现有设备条件能不能准确定量分析?,如果不能,有其他方法吗?[img=,690,499]https://ng1.17img.cn/bbsfiles/images/2022/04/202204231130009021_5249_3368413_3.png!w690x499.jpg[/img][img=,690,515]https://ng1.17img.cn/bbsfiles/images/2022/04/202204231130159976_4411_3368413_3.png!w690x515.jpg[/img][img=,690,521]https://ng1.17img.cn/bbsfiles/images/2022/04/202204231130325429_9731_3368413_3.png!w690x521.jpg[/img][img=,690,520]https://ng1.17img.cn/bbsfiles/images/2022/04/202204231130526089_7739_3368413_3.png!w690x520.jpg[/img]
我要开始做吡啶甲酸铬的测定,按照保健食品规范的标准测定,其中流动相的配制中有0.125mol/L磷酸盐缓冲液,说的不清楚。到底是用磷酸氢二钾和磷酸二氢钾怎么配置的?哪位大侠知道的话告知一下,不胜感激。
目前分析一个产品3-吡啶甲酸酐,CAS16837-38-0。需要测定其纯度。能找到的资料是熔点124℃,沸点200℃/1mmHg。可以用GC分析吗???这个产品是一个酸酐,请问各位有什么测定酸酐的好方法吗??(酸酐似乎遇水易分解)http://simg.instrument.com.cn/bbs/images/brow/em09509.gif
据欧盟委员会消息,欧委会于11月15日公布了(EU)No 1161/2011号法规,更新了可添加到食品中的矿物质名单,其中包括批准EDTA铁钠作为铁强化剂。 据了解,此次纳入合法矿物质名单中的物质包括: .磷酸亚铁铵(ferrous ammonium phosphate) .EDTA铁钠 .硫酸钠 .硫酸钾 .吡啶甲酸铬(chromium picolinate) 该法规将于公布之日起20天后生效。
据欧盟委员会消息,欧委会于11月15日公布了(EU)No 1161/2011号法规,更新了可添加到食品中的矿物质名单,其中包括批准EDTA铁钠作为铁强化剂。据了解,此次纳入合法矿物质名单中的物质包括:.磷酸亚铁铵(ferrous ammonium phosphate).EDTA铁钠.硫酸钠.硫酸钾.吡啶甲酸铬(chromium picolinate)该法规将于公布之日起20天后生效。
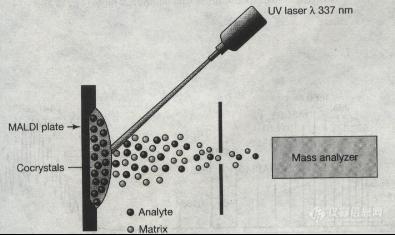
关于Maldi离子源Matrix-Assisted laser Desorption/IonizationMALDI源的出现解决了生物大分子的离子化难题,离子化过程与FBI离子源类似。1、使用基质,但基质为固体。2、MALDI用脉冲激光束轰击样品和基质的共结晶。对基质的要求是能吸收337nm紫外光并气化,能量由基质传给样品使样品一起气化并离子化。http://ng1.17img.cn/bbsfiles/images/2012/08/201208151035_383962_1978482_3.jpg一般MALDI常用的基质如下:1、α氰基-4羟基-肉桂酸 CCA 多肽2、3,5-二甲氧基-4-羟基肉桂酸 SA 蛋白3、龙胆酸(2,5-二羟基苯甲酸 DHB 聚合物4、吡啶甲酸 PA5、3-羟基吡啶甲酸 3HPAMALDI源由氮激光器产生短周期脉冲激光,产生的多为单电荷离子,效率很高,即使只有极少的样品也可分析优点1、质量数可达300,000Da。2、attomole 至femtomole级灵敏度。3、软电离方式,无或极少碎片离子。4、耐盐(样品含盐可达毫摩尔浓度)。5、适于分析复杂混合物缺点1、分辨率低。2、1000Da以下基质峰干扰。3、激光解吸附离子化有可能使样品光降解。4、串联质谱功能较弱,除非接反射装置进行源后衰变测量。5、不能分析非共价键相互作用。6、定量时需要内校准。7、如没有反射飞行装置,不能分析多肽修饰。8、对各种赋形剂的容忍度低(如含磷酸缓冲液,大于150mM的盐等。
最近单位新购一台[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url](安捷伦1200-6410,QQQ)在使用这台仪器进行蜂王浆中10-HDA和吡啶甲酸铬 的定量分析的时候发现,质谱结果和液相(安捷伦1100)结果相差非常大,达到数量级的差别。液相色谱的检验结果偏大并且多次检测结果很稳定;质谱结果则偏小,并且不稳定,具体如下:以吡啶甲酸铬德检测为例,我的标准系列分别为1.1 2.2 4.4ppm,第一次做标准曲线线性还不错有三个九,样品处理液有两个平行样,结果还平行,但是很低。然后大约30分钟以后,把1.1的那个标准溶液做为样品进行测定只有0.6,样品中含量为0。然后重新做标准系列,只有两个九。再大概十分钟后,将1.1的标准溶液重新做一个worklist,进样量分别设为1ul和5ul,发现1ul的峰更大。10-HDA在质谱上的检测结果也是类似情况。在液相色谱上检测时,只是将标准系列的浓度配的高了些,样品处理液用的是质谱上检测用的处理液。该蜂王浆样品多次在我们单位检测,在液相色谱上的结果与以前的检测结果基本一样。而[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]为新购仪器,用来定量还是第一次,对这两者如此大的区别,我们很是不解,不知道是不是我们在什么环节上没有注意到,请各位高手给予指点一二,多谢。
朋友们谁有苯甲酸甲酯、对甲苯磺酸、TEBAC、甲醇钠、4-二甲胺基吡啶、苯甲酰氯、乙硫醇、巴豆醛、乙酰乙酸甲酯、丙酰氯、DCP的国标、行标或企业标准啊?帮忙找一下啊,谢谢!
如果用顶空进样[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定对苯二甲酸中微量的吡啶,那标样应该怎么处理?这方面有没有相关的资料可以查?
大家好,不知道有没有研究过石墨炉做铅产生的光谱干扰问题?是283nm下做铅的石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]下,背景或光谱干扰??因为我之前做了一个吡啶甲酸铬中铅含量的研究,再看峰的时候,发现塞曼扣除的背景有点复杂,而且尾部很大,不过本人还真的没深入研究纵向塞曼扣背景的原理。 对铅的测量,有什么背景干扰?分子吸收干扰?以及光谱干扰?有没有可能Cr含量很高,而且是仲高温元素,在原子化过程中,阻挡了空心阴极灯的光,使其进入检测器减少,造成的假性吸收?还是由于热效应,使谱线变宽了,造成一下邻进吸收?
[color=#444444]现在用一种酯 2-甲基吡啶-4-甲酸乙酯做实验,做标准系列时,浓度大于30ppm,出峰效果挺好的,但是当浓度小于10ppm时,出峰效果很差,峰形不好,而用其他的吡啶酯不管什么浓度,出峰效果都很好,请问这种情况怎么解决,谢谢[/color]
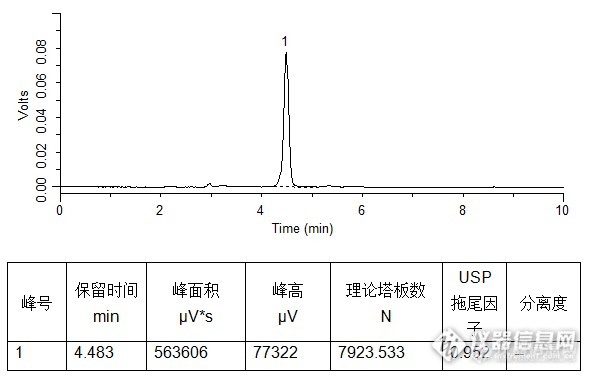
同一根柱子检测吡啶甲酸铬,20%甲醇水为流动相,截然不同的结果:http://simg.instrument.com.cn/bbs/images/brow/em39.gifhttp://simg.instrument.com.cn/bbs/images/brow/em44.gif图一:http://ng1.17img.cn/bbsfiles/images/2015/12/201512231349_579174_1987954_3.jpg换台仪器,相同流动相和样品,谱图如下:图二:http://ng1.17img.cn/bbsfiles/images/2015/12/201512231350_579175_1987954_3.png柱子没有问题,应该是仪器原因导致峰形拖尾,塔板数低。建议:1、 排查第一台仪器的单向阀和混合器;2、 按照流动相比例预先混合好后单泵使用(该建议治标不治本,最好把仪器捯饬好了再用);3、 重新连接柱子和仪器,避免死体积干扰。

【作者】 方奕珊; 李来生; 陈红; 张杨;【Author】 FANG Yi-shan,LI Lai-sheng,CHEN Hong,ZHANG Yang(The Centre of Analysis and Testing,Nanchang University,Nanchang 330047,China)【机构】 南昌大学分析测试中心;【摘要】 以4-二甲氨吡啶为内标,发展了一种高效液相色谱法,用于快速测定鸡蛋中氯羟吡啶的残留量。鸡蛋样品中的氯羟吡啶经乙腈匀浆提取,然后用碱性氧化铝柱富集,甲醇洗脱液浓缩后用流动相溶解残渣,0.45μm滤膜过滤后进样分析。色谱柱采用Platisil-C18柱(4.6mm×250mm,5μm),流动相为0.05mol.L-1甲酸铵-乙腈(85:15,v/v,pH 6.4),流速为0.8mL.min-1,二极管阵列定量检测波长为267nm,柱温25℃,进样量20μL。氯羟吡啶在0.5~80μg.mL-1范围内呈现良好的线性关系,线性方程为Y=0.254 4 X+0.703 9,相关系数为0.999 6。3个不同水平的标准添加平均回收率分别为85.3%,88.2%和83.7%,相对标准偏差≤2.25%(n=4)。最低检出限为1.8mg.kg-1。该方法快速、简便、准确、重现性好,适用于鸡蛋中兽药氯羟吡啶残留量的快速检测。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207301524_380581_2379123_3.jpg
二氯苯甲酸、乙酸等用什么色谱柱?样品中含二氯苯甲酸、乙酸、乙酸丁酯、醋酸铜、四甲基乙二胺、4-甲基吡啶应该怎么处理?用什么色谱柱?用安捷伦6890 FID色谱仪

小弟看不懂,特发过来和大家共同探讨。标准:《NY/T 916-2004 饲料添加剂 吡啶甲酸铬》,现行标准。该标准里,含量测定项下如下描述:http://ng1.17img.cn/bbsfiles/images/2014/01/201401101417_487409_2493208_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/01/201401101420_487411_2493208_3.jpg问题之一:前处理阶段,样品溶于20ml甲醇,超声30min后,取10ml蒸干,用10ml甲醇溶解后转移到50ml瓶中定容..........我想问:1》一开始共20ml溶液,超声30min后即使有盖子,由于甲醇的挥发性,此时总体积肯定少于20ml,为什么不写“补加甲醇至超声前重量”? 2》溶于甲醇后蒸干,然后又甲醇溶解,定容,如此折腾为哪般? 问题二:假设这个原料大约含量是100%,经过4.1.3.1处理后,供试品的待测液浓度应该是10μg/ml;而标准曲线的几个点,是0.025~0.02μg/ml,待测液严重超出标准曲线的线性范围,这样的方案设计合理吗? 我曾经尝试联系标准的起草人,没有得到答复。标准的起草人我查了下,来头还不小,为什么会出现这个情况呢..............我的理解不对?欢迎大家多多发表看法。
[table=100%][tr][td]用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]岛津色谱分析3-甲基吡啶 3-氰基吡啶 3-吡啶甲醛,乙醇为溶剂,得到的结果是吡啶甲醛和氰基吡啶的出锋时间相同,完全重合了,请问该怎样将他们分开?[/td][/tr][/table]
各位同仁!请教你们一个问题,我现在在做3-甲基吡啶和3-氰基吡啶的一个液相分析,目前我把4-个标品买回来了,分别是3-甲基吡啶,4-甲基吡啶,3-氰基吡啶,4-氰基吡啶;目前的一个情况就是3-甲基吡啶和4-甲基吡啶液相无法分开,3-氰基吡啶和4-氰基吡啶无法分开,液相打出来完全重合,我用的柱子是岛津C18柱子。流动相是甲醇:异丙醇:庚烷磺酸钠溶液=7:2:91,流速:1.0ml/min,检测波长261nm,请问有谁做过这样的液相分析,能否告诉小女子一下,万分感谢!
刚刚摸索用内标法测定2-氰基吡啶和3-氰基吡啶纯品的含量,不知道选哪种内标物比较好?(纯品中可能还含有甲苯、吡啶、2-甲基吡啶/3-甲/4-甲、4-氰基吡啶)看到一篇文献中以3-氰基吡啶为内标物测定2-氰基吡啶水溶液的含量,但以我们现在的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件,2-氰基吡啶和3-氰基吡啶的样品峰并不能完全分开,还有一小部分互溶,好像达不到内标法的要求。用甲醇或乙醇作内标物不知道合适不?期盼高手解答一下。不胜感激!
用吡啶和一氯甲烷反应,生成N-甲基吡啶一氯化物,可能有其它杂质(不知道是什么),怎么控制这个反应?
吡啶,有机化合物,是含有一个氮杂原子的六元杂环化合物。可以看做苯分子中的一个(CH)被N取代的化合物,故又称氮苯,无色或微黄色液体,有恶臭。吡啶及其同系物存在于骨焦油、煤焦油、煤气、页岩油、石油中。吡啶在工业上可用作变性剂、助染剂,以及合成一系列产品(包括药品、消毒剂、染料等)的原料。 英文名称: pyridine 中文名称2: 氮(杂)苯 CAS No.: 110-86-1 分子式: C5H5N 分子量: 79.10吡啶结构 吡啶的结构与苯非常相似,近代物理方法测得,吡啶分子中的碳碳键长为139pm,介于C-N单键 (147pm)和C=N双键(128pm)之间,而且其碳碳键与碳氮键的键长数值也相近,键角约为120°,这说明吡啶环上键的平均化程度较高,但没有苯完全。 吡啶环上的碳原子和氮原子均以sp2杂化轨道相互重叠形成σ键,构成一个平面六元环。每个原子上有一个p轨道垂直于环平面,每个p轨道中有一个电子,这些p轨道侧面重叠形成一个封闭的大π键,π电子数目为6,符合4n+2规则,与苯环类似。因此,吡啶具有一定的芳香性。氮原子上还有一个sp2杂化轨道没有参与成键,被一对孤对电子所占据,是吡啶具有碱性。吡啶环上的氮原子的电负性较大,对环上电子云密度分布有很大影响,使π电子云向氮原子上偏移,在氮原子周围电子云密度高,而环的其他部分电子云密度降低,尤其是邻、对位上降低显著。所以吡啶的芳香性比苯差。 在吡啶分子中,氮原子的作用类似于硝基苯的硝基,使其邻、对位上的电子云密度比苯环降低,间位则与苯环相近,这样,环上碳原子的电子云密度远远少于苯,因此象吡啶这类芳杂环又被称为“缺π”杂环。这类杂环表现在化学性质上是亲电取代反应变难,亲核取代反应变易,氧化反应变难,还原反应变易。吡啶性质 外观与性状: 无色或微黄色液体,有恶臭。 熔点(℃): -41.6 沸点(℃): 115.3 相对密度(水=1): 0.9827 折射率:1.5067(25℃) 相对蒸气密度(空气=1): 2.73 饱和蒸气压(kPa): 1.33/13.2℃ 闪点(℃): 17 引燃温度(℃): 482 爆炸上限%(V/V): 12.4 爆炸下限%(V/V): 1.7 偶极距:吡啶为极性分子,其分子极性比其饱和的化合物——哌啶大。这是因为在哌啶环中,氮原子 只有吸电子的诱导效应(-I),而在吡啶环中,氮原子既有吸电子的诱导效应,又有吸电子的共轭效应(-C)。 溶解性: 溶于水、醇、醚等多数有机溶剂。吡啶与水能以任何比例互溶,同时又能溶解大多数极性及非极性的有机化合物,甚至可以溶解某些无机盐类。所以吡啶是一个有广泛应用价值的溶剂。吡啶分子具有高水溶性的原因除了分子具有较大的极性外,还因为吡啶氮原子上的未共用电子对可以与水形成氢键。吡啶结构中的烃基使它与有机分子有相当的亲和力,所以可以溶解极性或非极性的有机化合物。而氮原子上的未共用电子对能与一些金属离子如Ag、Ni、Cu等形成配合物,而致使它可以溶解无机盐类。 与水形成共沸混合物,沸点92~93℃。(工业上利用这个性质来纯化吡啶。) 光谱性质: (1)吡啶的红外光谱(IR):芳杂环化合物的红外光谱与苯系化合物类似,在3070~3020cm-1处有C—H伸缩振动,在1600~1500cm-1有芳环的伸缩振动(骨架谱带),在900~700cm-1处还有芳氢的面外弯曲振动。 (2)吡啶的核磁共振氢谱(HNMR):吡啶的氢核化学位移与苯环氢(δ7.27)相比处于低场,化学位移大于7.27,其中与杂原子相邻碳上的氢的吸收峰更偏于低场。当杂环上连有供电子基团时,化学位移向高场移动,取代基为吸电性时,则化学位移向低场移动。 (3)吡啶的紫外吸收光谱(UV):吡啶有两条紫外光谱吸收带,一条在240~260nm(ε=2000),相应于π→π*跃迁(与苯相近)。另一条在270nm的区域,相应于n→π*跃迁(ε=450)。吡啶化学性质 吡啶及其衍生物比苯稳定,其反应性与硝基苯类似。典型的芳香族亲电取代反应发生在3、5位上,但反应性比苯低,一般不易发生硝化、卤化、磺化等反应。吡啶是一个弱的三级胺,在乙醇溶液内能与多种酸(如苦味酸或高氯酸等)形成不溶于水的盐。工业上使用的吡啶,约含1%的2-甲基吡啶,因此可以利用成盐性质的差别,把它和它的同系物分离。吡啶还能与多种金属离子形成结晶形的络合物。吡啶比苯容易还原,如在金属钠和乙醇的作用下还原成六氢吡啶(或称哌啶)。吡啶与过氧化氢反应,易被氧化成N-氧化吡啶。 (1)碱性和成盐 吡啶氮原子上的未共用电子对可接受质子而显碱性。吡啶的pKa为5.19,比氨(pKa9.24)和脂肪胺(pKa10~11)都弱。原因是吡啶中氮原子上的未共用电子对处于sp2杂化轨道中,其s轨道成分较sp3杂化轨道多,离原子核近,电子受核的束缚较强,给出电子的倾向较小,因而与质子结合较难,碱性较弱。但吡啶与芳胺(如苯胺,pKa4.6)相比,碱性稍强一些。 吡啶与强酸可以形成稳定的盐,某些结晶型盐可以用于分离、鉴定及精制工作中。吡啶的碱性在许多化学反应中用于催化剂脱酸剂,由于吡啶在水中和有机溶剂中的良好溶解性,所以它的催化作用常常是一些无机碱无法达到的。 吡啶不但可与强酸成盐,还可以与路易斯酸成盐。 此外,吡啶还具有叔胺的某些性质,可与卤代烃反应生成季铵盐,也可与酰卤反应成盐。 (2)亲电取代反应 吡啶是“缺π”杂环,环上电子云密度比苯低,因此其亲电取代反应的活性也比苯低,与硝基苯相当。由于环上氮原子的钝化作用,使亲电取代反应的条件比较苛刻,且产率较低,取代基主要进入3(β)位。 与苯相比,吡啶环亲电取代反应变难,而且取代基主要进入3(β)位,可以通过中间体的相对稳定性来说明这一作用。 由于吸电性氮原子的存在,中间体正离子都不如苯取代的相应中间体稳定,所以,吡啶的亲电取代反应比苯难。比较亲电试剂进攻的位置可以看出,当进攻2(α)位和4(γ)位时,形成的中间体有一个共振极限式是正电荷在电负性较大的氮原子上,这种极限式极不稳定,而3(β)位取代的中间体没有这个极不稳定的极限式存在,其中间体要比进攻2位和4位的中间体稳定。所以,3位的取代产物容易生成。 (3)亲核取代反应 由于吡啶环上氮原子的吸电子作用,环上碳原子的电子云密度降低,尤其在2位和4位上的电子云密度更低,因而环上的亲核取代反应容易发生,取代反应主要发生在2位和4位上。 吡啶与氨基钠反应生成2-氨基吡啶的反应称为齐齐巴宾(Chichibabin)反应,如果2 位已经被占据,则反应发生4位,得到4-氨基吡啶,但产率低。如果在吡啶环的α位或γ位存在着较好的离去基团(如卤素、硝基)时,则很容易发生亲核取代反应。如吡啶可以与氨(或胺)、烷氧化物、水等较弱的亲核试剂发生亲核取代反应。 (4)氧化还原反应 由于吡啶环上的电子云密度低,一般不易被氧化,尤其在酸性条件下,吡啶成盐后氮原子上带有正电荷,吸电子的诱导效应加强,使环上电子云密度更低,更增加了对氧化剂的稳定性。当吡啶环带有侧链时,则发生侧链的氧化反应。 吡啶在特殊氧化条件下可发生类似叔胺的氧化反应,生成N-氧化物。例如吡啶与过氧酸或过氧化氢作用时,可得到吡啶N-氧化物。 吡啶N-氧化物可以还原脱去氧。在吡啶N-氧化物中,氧原子上的未共用电子对可与芳香大π键发生供电子的p-π共轭作用,使环上电子云密度升高,其中α位和γ位增加显著,使吡啶环亲电取代反应容易发生。又由于生成吡啶N-氧化物后,氮原子上带有正电荷,吸电子的诱导效应增加,使α位的电子云密度有所降低,因此,亲电取代反应主要发生在4(γ)上。同时,吡啶N-氧化物也容易发生亲核取代反应。 与氧化反应相反,吡啶环比苯环容易发生加氢还原反应,用催化加氢和化学试剂都可以还原。 吡啶的还原产物为六氢吡啶(哌啶),具有仲胺的性质,碱性比吡啶强(pKa11.2),沸点106℃。很多天然产物具有此环系,是常用的有机碱。 (5)环上取代基与母环的影响 取代基对水溶解度的影响:当吡啶环上连有-OH、-NH2后,其衍生物的水溶度明显降低。而且连有-OH、-NH2数目越多,水溶解度越小。. 其原因是吡啶环上的氮原子与羟基或氨基上的氢形成了氢键,阻碍了与水分子的缔合。取代基对碱性的影响:当吡啶环上连有供电基时,吡啶环的碱性增加,连有吸电基时,则碱性降低。与取代苯胺影响规律相似。吡啶应用 除作溶剂外,吡啶在工业上还可用作变性剂、助染剂,以及合成一系列产品(包括药品、消毒剂、染料、食品调味料、粘合剂、炸药等)的起始物。 吡啶还可以用做催化剂,但用量不可过多,否则影响产品质量。吡啶来源 吡啶可从天然煤焦油中获得,也可由乙醛和氨制得。吡啶及其衍生物也可通过多种方法合成,其中应用最广的是汉奇吡啶合成法,这是用两分子的β-羰基化合物,如乙酰乙酸乙酯与一分子乙醛缩合,产物再与一分子的乙酰乙酸乙酯和氨缩合形成二氢吡啶化合物,







 我要推广仪器
我要推广仪器
 下载APP
下载APP