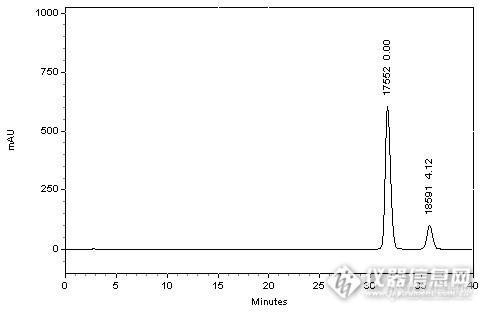
http://ng1.17img.cn/bbsfiles/images/2014/09/201409171441_514220_2222981_3.jpg*注:峰上标由下至上依次为理论塔板数和分离度。 HPLC Conditions :色谱柱:CAPCELL PAK C18 MGII S5; 4.6mm i.d.×250mm 流动相:三乙胺磷酸溶液【三乙胺溶液(1→100),用磷酸调节pH至4.3】/乙腈=87/13流速: 1000µL / min温度: 30°C检测: UV 325nm进样量: 10 µL分析结果与讨论 本实验依据国家标准分离加替沙星与N-甲基加替沙星,实验中分别尝试了不同规格的CAPCELL PAK C18 系列MG、MGII、AQ色谱柱,发现MGII色谱柱可得到最好分离结果。实验结果如上图所示,使用MGII色谱柱进行分析时,加替沙星峰理论塔板数达到17552,且加替沙星与N-甲基加替沙星峰分离度为4.12,已达到2013年所起草国家标准征求意见稿的分离度大于4的要求。
领导希望能探索出一个流动相,可以对辛伐他汀,辛伐他汀铵盐,盐酸环丙沙星,环丙羧酸用一种流动相进行预检,以确定其有关物质情况,有没有人做过这方面的研究,求帮助
[B]动物源性食品中四环素、沙星类[/B] 残留量的快速测定方法1 范围本方法规定了动物源性食品中四环素类、沙星类高效液相色谱的快速测定方法。本方法适用于动物源性食品中四环素类、沙星类高效液相色谱的快速测定。2.1 原理试样中的残留物经四环素类、沙星类快速检测前处理试剂盒处理,样液经四环素类专用层析柱净化、浓缩用高效液相色谱检测,外标法定量。2.2 试剂和材料除另有规定外,所有试剂均为分析纯,水为重蒸馏水。2.2.1 乙腈:色谱纯。2.2.2 甲醇:色谱纯。2.2.3 三乙胺(分析纯)2.2.4 磷酸(85%)(分析纯)2.2.5 磷酸氢二钠:优级纯。2.2.6 乙二胺四乙酸二钠。2.2.7 柠檬酸:分析纯。2.2.8 磷酸氢二钠溶液:0.2mol/L。称取28.41g磷酸氢二钠,用水溶解,定容至1000mL。2.2.9 柠檬酸溶液:0.1mol/L。称取21.01g柠檬酸,用水溶解,定溶至1000mL。2.2.10 Mcllvaine缓冲溶液:将1000mL0.1mol/L柠檬酸溶液与625mL0.2mol/L磷酸氢二钠溶液混合。2.2.11 Na2EDTA-Mcllvaine缓冲溶液:0.1mol/L。称取60.5g乙二胺四乙酸二钠放入1625mLMclllvaine缓冲溶液中,使其溶解,摇匀。2.2.12 标准品: 土霉素、四环素、金霉素、强力霉素、氧氟沙星、诺氟沙星、环丙沙星、单诺沙星、恩诺沙星纯度大于98 %。2.2.13 标准贮备溶液:分别称取土霉素、四环素、金霉素、强力霉素、氧氟沙星、诺氟沙星、环丙沙星、单诺沙星、恩诺沙星各10mg,用甲醇溶解并定溶于100 mL棕色容量瓶中,配制成100 µ g/mL的标准贮备液,置于-20℃保存,有效期三个月。2.2.14 混合标准工作溶液:用流动相稀释标准贮备溶液,配制成土霉素、四环素、金霉素、强力霉素、氧氟沙星、诺氟沙星、环丙沙星、单诺沙星、恩诺沙星均为10 µ g/mL的混合标准溶液。0~4℃避光保存。2.2.15 四环素、沙星类快速检测前处理试剂盒*。2.3 仪器和设备2.3.1 高效液相色谱仪:配紫外-可见光波长检测器。2.3.2 匀浆机。2.3.3 固相萃取机2.3.4 离心机:4000 r/min。2.3.5 调速多用振荡器。2.3.6 聚四氟乙烯离心管: 2.5 mL,50 mL,具塞。2.4 样品制备准确称取已捣碎的样品5.00 g于50 mL离心管中,先加入四环素、沙星类快速检测前处理试剂盒中的提取剂 (液体20mL),用调速多用振荡器150 rpm振荡3 min,,4000 r/min离心5 min,收集上清液10mL加入四环素专用层析柱中(使用前依次用5mL甲醇、5mL提取剂、5mL水激活)挤干,用2mL提取剂洗涤,用0.80mL甲醇洗脱,收集洗脱液,用0.2mL流动相定容至1.0mL。供仪器测定。2.5 测定2.5.1 液相色谱条件a) 色谱柱: C18柱,250 mm×4 mm(i.d.),粒度5µ m b) 流动相: 0.05 mol/L磷酸/三乙胺缓冲液(pH2.4)+乙腈(80+20,V/V) c) 流速: 1.0mL/min d) 柱温: 室温 e) 检测波长: 四环素类紫外检测器350 nm。沙星类 紫外检测器310nm;荧光检测器激发波长280nm,发射波长450nm。f) 进样量:50 uL。3.5.2 标准工作曲线绘制移取各移取四环素类、沙星类混和标准液,用流动相稀释成20 ng/mL、50 ng/mL、250 ng/mL、500 ng/mL标准工作溶液。按液相色谱条件(3.5.1)进行测定,以色谱峰的峰面积为纵坐标,与其对应的浓度为横坐标作图,绘制标准工作曲线,标准工作曲线范围:20.0~500 ng/mL。 3.5.3 试样测定 用微量进样器准确吸取试样溶液(3.4),按液相色谱条件(3.5.1)进行测定,记录色谱峰的保留时间和峰面积。3.6 结果计算按式(1)分别计算供试样品中的四环素类、沙星类残留量。 2×ci×Vω= …… (1)mω-水产品中四环素、沙星类残留量,μg/kg;ci -标准曲线上查出试样溶液中四环素、沙星类标准工作溶液的浓度,(μg/L);V-最终定容体积数,mL;2-换算常数;m-供试试料样品重量,g。本方法分别计算四环素类、沙星类结果。3.7 检测限本方法土霉素、四环素检测限为20µ g/kg;金霉素、强力霉素的检测限为50µ g/kg,沙星类为:5µ g/kg3.8 回收率 本方法土霉素、四环素、金霉素、强力霉素回收率为:75%~85%;沙星类回收率为:75%~85%相关谱图附件可见联 系 人:王 伦 手 机:13810239506 EMAIL:wwj613@sina.com
要检测肉里的兽药残留沙拉沙星,标准品有沙拉沙星和盐酸沙拉沙星,一个2百多一个三千多,这2种标准品有什么区别
大家好: 我们的养殖场使用的氧氟沙星我们却在蛋中检测出诺氟沙星请大家分析一下原因,我们是用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]检测的。谢谢!
[font=宋体][font=宋体]检测农产品中的喹诺酮类药物对于第三方检测食品实验室而言是再熟悉不过的项目了,氧氟沙星作为常检项目之一,主要针对水产品、牛羊等畜肉,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]法检测主要依据国家标准[/font][font=Calibri]GB 31658.17-2021[/font][font=宋体]和农业部[/font][font=Calibri]1077[/font][font=宋体]号公告[/font][font=Calibri]-1-2008[/font][font=宋体]。就这么一个常规项目,在前段时间出现诡异的污染现象。[/font][/font][font=宋体]当时在做一批的水产品检测时,发现[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]检测结果所有的样品中都有氧氟沙星检出,而且结果均超过了最大残留限值。当时首先考虑的是基质干扰,因为氧氟沙星常出现保留时间接近的干扰峰,查看相对离子丰度比发现确实是氧氟沙星。考虑到空白样品中也有氧氟沙星检出,可能是前处理过程中带入的污染。重新配置了内标工作溶液,并将这批样品换人重做后问题并没有得到解决,因此怀疑污染源在仪器端。[/font][font=宋体][font=宋体]我将第一次处理的样品换到另一台[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]上检测,结果也有氧氟沙星检出,检出值小于之前的结果。分析三次的测试结果发现:所有样品、溶剂空白、样品空白都有氧氟沙星检出,保留时间基本一致,同一台仪器的仪器不同样品之间的结果接近,同一样品不同仪器的检测结果不一致。怀疑污染是在进样过程中引入,连同容器一起更换了新的洗针溶液,充分灌注洗针液通道,多次运行清洗进样针、针密封清洗、注射器清洗程序。用之前的溶剂空白,连续进样[/font][font=Calibri]10[/font][font=宋体]次,测试结果显示,溶剂空白中不再有氧氟沙星检出。将之前换下的洗针溶液取样上机测试,发现两台仪器的弱洗溶液中都有氧氟沙星检出。将之前的水产样品上机测试,结果均未检出氧氟沙星。[/font][/font][font=宋体]在后续查找污染原因时发现,同时在配置洗针溶液时未规范佩戴手套与口罩,同时他最近在服用氧氟沙星片剂。当时配置的洗针液直接添加到仪器洗针液的容器中,剩余部分倒入了另一台仪器的洗针液容器中。[/font][font=宋体]这次污染事件提醒了我们,在使用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]这类高灵敏度的仪器时,规范的实验操作的重要性。实验室防护品不仅保护我们的健康,也是实验结果准确的保障。不同仪器间,避免出现试剂混用的现象,这样当出现问题时,我们才能更准确的找到原因。[/font]

[align=center][color=#000000]环丙沙星的USP方法分析[/color][/align][align=left][color=#000000]环丙沙星为合成的第三代喹诺酮类抗菌药物,具广谱抗菌活性,杀菌效果好,几乎对所有细菌的抗菌活性均较诺氟沙星及依诺沙星强2~4倍,对肠杆菌、绿脓杆菌、流感嗜血杆菌、淋球菌、链球菌、军团菌、金黄色葡萄球菌具有抗菌作用。[/color][color=#000000]实验室对客户提供的环丙沙星专属性溶液(0.35mg/mL,以环丙沙星浓度计)以及系统适用性溶液(7.5ug/mL,以环丙沙星浓度计,由专属性溶液稀释得到)在四款C18色谱柱(ODS、MG、AQ-S5、AQ-S3)上分别进行分析考察实验。[/color]通过实验,比较杂质B与C、杂质C与主峰、主峰与杂质I之间的分离度,以及主峰拖尾因子情况。[color=#2b4c72][color=#3e3e3e][color=#000000]专属性溶液分析对比结果如表1所示,在专属性溶液分析中,主峰浓度较大产生过载,对主峰前后杂质分离度影响较大,但四款色谱柱均满足分离度≥6.0的要求。其中,客户主要关注杂质C和主峰分离情况,其中[b]AQ-S3[/b]色谱柱得到最好分离,分离度为[b]7.786[/b]。[/color][/color][/color][/align][align=left][/align][align=center][color=#2b4c72][color=#3e3e3e][color=#3e3e3e]表1 专属性溶液分离比较表[/color][/color][/color][/align][align=center][color=#2b4c72][color=#3e3e3e][color=#3e3e3e][img=,690,161]http://ng1.17img.cn/bbsfiles/images/2017/05/201705180842_01_2222981_3.png[/img][/color][/color][/color][/align]在系统适用性溶液分析中,四款色谱柱所得结果均符合USP要求,即拖尾因子≤2.0,杂质C和主峰之间分离度≥6.0。AQ-S3色谱柱虽然得到了杂质C和主峰的最好分离,但对于杂质B和C以及主峰和主峰后杂质I来讲分离不够理想;MG色谱柱体现出了整体最好分离,结果如表2所示。[align=center][color=#3e3e3e]表2 系统适用性分离比较表[/color][/align][align=center][color=#3e3e3e][img=,690,168]http://ng1.17img.cn/bbsfiles/images/2017/05/201705180842_02_2222981_3.png[/img][/color][/align][align=left][color=#3e3e3e][/color][color=#3e3e3e]图1、图2为AQ-S3柱分析专属性溶液和系统适用性谱图,图3、图4为MG柱相应分析谱图。[/color][/align][align=center][color=#3e3e3e][img=,690,218]http://ng1.17img.cn/bbsfiles/images/2017/05/201705180842_04_2222981_3.png[/img][/color][/align][align=center][color=#3e3e3e]图1 AQ-S3色谱柱专属性分析色谱图[/color][/align][align=center][img=,690,221]http://ng1.17img.cn/bbsfiles/images/2017/05/201705180842_05_2222981_3.png[/img][/align][align=center][color=#3e3e3e]图2 AQ-S3色谱柱系统适用性分析色谱图[/color][/align][align=center][color=#3e3e3e][img=,690,224]http://ng1.17img.cn/bbsfiles/images/2017/05/201705180842_06_2222981_3.png[/img][/color][/align][align=center][color=#3e3e3e]图3 MG专属性分析色谱图[/color][/align][align=center][color=#3e3e3e][img=,690,222]http://ng1.17img.cn/bbsfiles/images/2017/05/201705180842_07_2222981_3.png[/img][/color][/align][align=center][color=#3e3e3e][color=#3e3e3e]图4 MG系统适用性分析色谱图[/color][/color][color=#3e3e3e][color=#3e3e3e][/color][/color][/align][align=left][color=#3e3e3e][color=#3e3e3e][img=,690,166]http://ng1.17img.cn/bbsfiles/images/2017/05/201705180842_03_2222981_3.png[/img][/color][/color][/align][align=left][color=#3e3e3e]*注:S3和S5分别代表色谱柱填料粒径为3um和5um,MG和ODS规格均为S5。[/color][/align]
请教各位前辈:1.听说有个ZN1009-2004标准,能检测水产品中,哪位前辈有,能否给我邮一份?2.有其他能同时前处理和检测水产品中恩诺沙星、环丙沙星、诺氟沙星、氧氟沙星的方法吗?QQ:3733818E-mail:yardin@126.com谢了先!!
我自己翻了,希望能有更专业的帮忙翻译,我自己对照学习氧 氟 沙 星 片Yangfushaxing PianOfloxacin Tablets本品含氧氟沙星(C18H20FN3O4)应为标示量的90.0%~110.0%。【性状】 本品为类白色或微黄色片或薄膜衣片,除去包衣后显类白色至微黄色。【鉴别】 (1)在含量测定项下记录的色谱图中,供试品溶液主峰的保留时间应与对照品溶液主峰的保留时间一致。(2)取本品细粉适量,用0.1mol/L盐酸溶液溶解并稀释制成每1ml中含氧氟沙星6μg的溶液,滤过,滤液照紫外-可见分光光度法(附录Ⅳ A)测定,在294nm的波长处有最大吸收。【检查】 有关物质 取含量测定项下的供试品贮备液作为供试品溶液;精密量取适量,加0.1mol/L盐酸溶液稀释成每1ml中含6μg的溶液,作为对照溶液。照氧氟沙星有关物质项下的方法测定,供试品色谱图中如有杂质峰,单个杂质峰面积不得大于对照溶液主峰面积(0.5%),各杂质峰面积的和(任何小于对照溶液主峰面积的0.05倍的峰可忽略不计)不得大于对照溶液主峰面积的2倍(1.0%)。溶出度 取本品,照溶出度测定法(附录Ⅹ C 第一法),以盐酸溶液(9→1000)900ml为溶出介质,转速为每分钟50转,依法操作,经30分钟时,取溶液适量,滤过,精密量取续滤液2ml,置50ml量瓶中,加溶出介质稀释至刻度,摇匀,照紫外-可见分光光度法(附录Ⅳ A),在294nm的波长处测定吸收度;另取氧氟沙星对照品适量,精密称定,加溶出介质溶解并稀释成每1ml中约含4.5μg的溶液,同法测定,计算每片的溶出量。限度为标示量的80%,应符合规定。其他 应符合片剂项下有关的各项规定(附录Ⅰ A)。【含量测定】照高效液相色谱法(附录Ⅴ D)测定。色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;以醋酸铵高氯酸钠溶液(取醋酸铵4.0g和高氯酸钠7.0g,加水1300使溶解,用磷酸调节pH值至2.2)-乙腈(85:15)为流动相;检测波长为294nm。取氧氟沙星适量,用0.1mol/L盐酸溶液溶解并稀释制成每1ml中约含1mg的溶液,置紫外光灯(254nm)下照射4小时以上,取10μl注入液相色谱仪,理论板数按氧氟沙星峰计算不低于5000,紧邻氧氟沙星峰前的杂质峰与氧氟沙星峰的分离度应符合要求。测定法 取本品10片,精密称定,研细,精密称取适量(约相当于氧氟沙星0.1g),置100ml量瓶中,加0.1mol/L盐酸溶液溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试品贮备液,精密量取5ml,置50ml量瓶中,加0.1mol/L盐酸溶液至刻度,摇匀,精密量取10μl分别注入液相色谱仪,记录色谱图;另取氧氟沙星对照品适量,精密称定,用0.1mol/L盐酸溶液溶解并定量稀释制成每1ml中含氧氟沙星0.12mg的溶 液,同法测定,按外标法以峰面积计算,即得。【类别】 同氧氟沙星。【规格】 0.1g【贮藏】 遮光,密封保存。
最近收到一批加替沙星样品(原料药)要求用[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测定铁的含量,请问哪位高手预处理应该怎么进行啊?以前没有做过,连一点参考也没有,急。。。谢谢。[em09501]
问题: 各位亲,西诺沙星纯品用什么溶解?
我在做畜产品中恩诺沙星时,用的是荧光检测器,刚配的标样为啥出两个峰呢?一个在2点几,一个在三点几?是仪器出什么问题了吗?还有做畜产品中伊维菌素时出峰更奇怪了,我设定停止时间是20分钟,他就在20分钟时出峰,设定在25分钟,他就在25分钟出峰,太郁闷了!请高手指点!谢谢!
我配制的恩诺沙星标准溶液,荧光检测器分析后总出现环丙沙星的峰,是买的标准品不纯(Dr公司)还是我配制时有污染,请老师指教。

国家局下发加替沙星胶囊的质量标准,要求加替沙星胶囊含量检测时,加替沙星与甲基加替沙星的分离度应达到8.0。我们用了很多柱子都没有办法解决。除了改变pH在4.7时,分离度可以在10。 哪位高手有办法,在不改变质量标准的前提下,将分离度达到要求。
各位大侠,有没有做过水产品中恩诺沙星、诺氟沙星和环丙沙星残留的快速筛选测定 用的是胶体金免疫渗滤法
农业部235号里有恩诺沙星的限量,没有环丙沙星的限量,实验中恩诺沙星是以恩诺沙星和环丙沙星之和计的,检测是测的两个沙星的和,限量直接写恩诺沙星的限量吗
[color=#444444]大家好,我想问个问题,在食品检测恩诺沙星时,报告判定时是以恩诺沙星和环丙沙星之和计,这是为什么?跟检测样品有关还是什么原因?请大神赐教![/color]
请教牛奶中氟喹诺酮残留的样品前处理方法,是SPE提取,用的是lc/ms测定,主要是氧氟沙星,诺氟沙星,环丙沙星,恩诺沙星,洛美沙星!
建立一种快速测定恩诺沙星的新方法。利用鲁米诺- 铁氰化钾化学发光体系,结合流动注射技术,对恩诺沙星进行测定。实验结果表明,恩诺沙星对鲁米诺- 铁氰化钾化学发光反应具有显著的增敏作用,在最佳实验条件下,该方法的线性范围为6 × 10-11~6 × 10-7mol/L,检出限(3σ)为2.7 × 10-13mol/L,RSD 为1.2%(n=11)。该方法用于注射液中恩诺沙星含量的测定,结果令人满意。
大家好: 我在做食品中沙星检测的时候,发现氧氟沙星、诺氟沙星怎么改变流动相比例都不能完全分离,甚至重合在一块,甚是头疼,请大家帮忙。
1.概述环丙沙星酶联免疫反应测试盒是利用竞争性的酶联反应原理,用于饲料、肉类组织(牛肉、鸡肉和猪肉)、鱼虾、牛奶、组织、血清和尿液中环丙沙星残留的定量检测。该试剂盒具有以下特点:Ø 快速,高回收率(75-95%),多种样品的低成本提取方法。Ø 高灵敏度(0.35ng/g或ppb),低检测下限(饲料有机提取法0.525ng/g或ppb)。Ø 高重复性。Ø 检测过程只需要不到1.5小时。 2.试剂盒原理环丙沙星酶联免疫反应测试盒基于竞争性酶联反应原理,含有环丙沙星抗体的药物已经包被于微孔板上。药物分析时,样品同HRP酶标记物共同被添加到板孔中。如果样品中含有环丙沙星,会竞争包被抗体,抑制HRP酶标记物与板上包被的抗体结合。加入底物后,产物的颜色强弱与样品中药物的浓度成反比。
莫西沙星混合对照品究竟有几个成分?5个还是包括莫西沙星在内的6个?Dissolve 5 mg of moxifloxacin for peak identification CRS (containing impurities A, B, C, D and E) in solution A and dilute to 5.0 ml with the same solution.
我最近做沙星,空白样品添加诺氟沙星和环丙沙星出不来,氧氟沙星和恩诺沙星没问题。纯标品进样也没问题。各位大侠帮我分析分析可能哪里出了问题!! 现在我们实验室的室温挺低的也就有6-7度,会有影响吗?http://simg.instrument.com.cn/bbs/images/default/emyc1010.gif
实验室采用3.4ml磷酸 三乙胺调pH=2.4可是这样的话,走混标工作液的时候,诺氟沙星和氧氟沙星是分不开的,怎么办?? 有谁知道?
1.概述REAGEN™恩诺沙星酶联免疫反应测试盒是利用竞争性的酶联反应原理,用于饲料、肉类组织(牛肉、鸡肉和猪肉)、鱼虾、牛奶、组织、血清和尿液中恩诺沙星残留的定量检测。该试剂盒具有以下特点:Ø 快速,高回收率(75-105%),多种样品的低成本提取方法。Ø 高灵敏度(0.1ng/g或ppb),低检测下限(牛奶0.5ng/g或ppb)。Ø 高重复性。Ø 检测过程只需要不到1.5小时。 2.试剂盒原理REAGEN™恩诺沙星酶联免疫反应测试盒基于竞争性酶联反应原理,含有恩诺沙星的抗原已经包被于微孔板上。药物分析时,样品同特异性一抗共同被添加到板孔中。如果样品中含有药物,会竞争一抗,抑制抗体与板上包被的药物抗原结合。加入酶标记的二抗,形成包被抗原-抗体-酶标二抗复合物。加入底物后,产物的颜色强弱与样品中药物的浓度成反比。
左氧氟沙星和左氧氟沙星酯2个性质很近的物质如何使得他们有很好的分离度希望有做过的,知道的,懂得的,不吝赐教!十分感谢!
检测一包猪肉冻干粉,里面添加了环丙沙星和恩诺沙星,添加量是150ppb,我检测130ppb,同时跟做的回收率是70%如果除回收检测结果就变成了185ppb,这种情况报哪种合适呢?求高人指点
恩诺沙星标准品是带硫酸盐的,带了盐的目标物最后的离子跟恩诺沙星本体的离子是一样的么
想问大家有没有做过鱼粉中的环丙沙星恩诺沙星,大概怎么提取的?直接称鱼粉吗还是要加点水啊?
[align=center]热脱附-[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]质谱分析热环境下汽车沙发释放的挥发性有机物[/align] 摘 要 汽车沙发部件在热环境中,会释放出许多挥发性有机物(VOC),污染环境,危害人体健康。本研究将汽车沙发放置在2 m3 特制塑料采样袋中,在热环境下释放挥发性有机物,然后以Tenax 管富集有机物,用热脱附-[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]质谱进行分析。结果表明:汽车沙发在热环境状态下,挥发出大量有机物,共定性检测出49 种挥发性有机物(VOC),包括11 种烷烃(7.46% )、13 种芳香类化合物(6.02% )、5 种醇醚类化合物(10.33% )、5 种酮类化合物(3.56% )、7 种酯类化合物(29.51% )、2 种醛类化合物(0.50% )、2 种含氮类化合物(29.36% )及4 种硅氧烷类化合物(5.51% )。通过将释放的挥发性有机物量换算成车内空气污染浓度可见,一个副驾驶位沙发释放的挥发性有机物对车内空气污染贡献浓度达7.36 mg/m3 ,是国家室内限值标准的12 倍,污染十分严重。 1 引 言 目前,汽车已成为人们生活的重要交通工具,车内的空气污染已对人体健康产生影响。车内挥发性有机物(VOC)主要来源于车内装饰材料,其中汽车沙发是主要来源之一。这些有害物质会致乘车人头晕、恶心、打喷嚏,甚至引起更严重的疾病,如孕妇流产、儿童的白血病及男性不育等。 2 实验部分 2.1 仪器与试剂 6890N/5975C 型[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]质谱联用仪(美国Agilient 公司) Master TD 型二次热脱附仪(意大利DANI 公司) 半导体冷阱捕集器 小轿车副驾驶位皮制沙发部件(由某汽车厂家提供,为新生产的、放置时间不超过30 d、即将安装在新车副驾驶位的沙发) 2 m3 塑料袋(日本三菱化学公司提供的特制塑料薄膜,在75 ℃下烘烤2 h 后加工制作成2 m3 的塑料袋) Tenax-TA 采样管(石英玻璃管,外径6mm,内装Tenax-TA150 mg,临用前在300 ℃下活化1 h)。10,100,500 和1000 mg/L 标准物质系列(甲苯、乙苯、对二甲苯、间二甲苯、苯乙烯、邻二甲苯及正十四烷混合标样,日本) 2.2 仪器参数 2.2.1 色谱条件 HP-1 色谱柱(60 m×0.32 mm×1.0 mm)弹性石英毛细管柱,载气为氦气,进样口温度为280 ℃,色谱柱升温条件为初始温度50 ℃,恒温4 min,以4 ℃/min 升温速率升到150 ℃,然后以10 ℃/min 升至300 ℃,恒温10 min。采用分流进样,分流比20∶1。 2.2.2 质谱条件 电离方式为EI,电子能量为70 eV,离子源温度230 ℃,MS 四极杆温度150 ℃,氦气 流速为1.4 mL/min 扫描范围为m/z 35~400。 2.2.3 热脱附条件 一次解析温度300 ℃,解析时间20 min。 2.2.4 半导体冷阱捕集器 填料Tenax TA,捕集温度0 ℃,解析温度300 ℃,解析时间10 min。 2.3 样品收集 取汽车沙发样品放置在2 m3 塑料袋中,将塑料袋口加热密封,再用无油真空泵抽其真空后,充1600 L 高纯度干燥空气,然后将放置样品的充气塑胶袋转放在24 m3 的温度为40 ℃试验箱内,在4.5 h后用活化后的Tenax 管采样,采样速率0.2 L/min,采样体积3 L,采样后用热脱附-[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]质谱联用仪进行分析。同时进行空白实验。 2.4 分析步骤 将采集好的吸附管,按照热脱附仪的操作要求,放置在自动热脱附托盘上。进行热脱附和分析。取5 支经老化的Tenax-TA 吸附管,用10 mL 微量注射器分别吸取标准系列溶液1 mL,经液体标准配气装置进入吸附管中,制成0,10,100,500 和1000 ng 标准系列管。经热脱附-[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]质谱分析,通过质谱峰进行定性与定量分析,以含量为横坐标,峰面积为纵坐标,绘制标准曲线。 3 结果与讨论 3.1 挥发性有机物的定性分析 图1 为小汽车沙发部件在热干燥环境下释放的挥发性有机物的总离子流色谱图,共出现53 个峰。对离子流色谱图进行NIST 05 标准谱图检索,配合人工图谱解析和查阅相关资料,同时结合色谱保留时间进行定性分析,鉴别出49 个峰,占总峰面积的92.24% 。它们分别属于包括11 种烷烃(7.46% ), 13 种芳香类化合物(6.02% ), 5 种醇醚类化合物(10.33% ), 5 种酮类化合物(3.56% ), 7 种酯类化合物(29.51% ), 2 种醛类化合物(0.50% ), 2 种含氮类化合物(29.36% ), 4 种硅氧烷类化合物(5.51% )及4 种未知物(7.75% )。[align=center][img]http://img.jgvogel.cn/2013/0403/0859153597.png[/img][/align] 3.2 挥发性有机物的定量分析 采用外标法对甲苯、乙苯、对/间二甲苯、苯乙烯、邻二甲苯及正十四烷进行定量分析,对于总挥发性有机物,从正己烷到正十六烷之间尽可能多的峰面积进行积分,其它组分的定量以甲苯的响应系数计算,从定量结果可以看出,汽车沙发在热干燥环境条件下总挥发性有机物(TVOCs)释放量为14.7 mg,其中释放最多的挥发性有机物是N,N-二甲基甲酰胺,其释放量为4.91 mg,占总挥发性有机物的33.4% 其次是酯类化合物,包括乙酸乙酯、丙烯碳酸酯、3-乙氧基丙酸乙酯、丁二酸二甲酯、戊二酸二甲酯、己二酸二甲酯、2,2,4-三甲基戊二醇异丁酯,合计释放量为5.03 mg,占总挥发性有机物的34.2% 芳香烃类化合物合计释放量为1.03 mg,占总挥发性有机物的7.0% 。 通过将释放的挥发性有机物量换算成车内空气浓度(以小汽车内部空间以2 m3 计算),从表1 可见,汽车沙发释放出的挥发性有机物对汽车空气污染的贡献较大。由于目前我国尚未制定专门的车内空[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]量标准,参考车内空气中挥发性有机物浓度要求(国家汽车浓度限值标准征求意见稿)及国家室内空气环境质量标准[10,11]的规定,汽车沙发释放出的苯、甲苯、二甲苯、乙苯及苯乙烯造成车内空气污染贡献浓度远低于车内空气中挥发性有机物浓度要求,但是总挥发性有机物对汽车内空[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]量的贡献浓度达7.36 mg/m3,是国家室内限值标准的12 倍(国家室内空气中总挥发性有机物浓度限值为0.60 mg/m3 )。可以看出,1 个副驾驶位的汽车沙发在热干燥环境下挥发性有机物的释放量很大。[img]http://img.jgvogel.cn/2013/0403/0859483625.png[/img][img]http://img.jgvogel.cn/2013/0403/0900187306.png[/img]3.3 结论 实验结果表明,在热干燥环境下,小汽车沙发制品释放出的挥发性有机物共得到53 个峰,从中鉴定出49 种挥发性有机物,其峰面积占总峰面积的92.24% 。汽车沙发释放的总挥发性有机物14.7 mg,对车内空气污染贡献浓度达7.36 mg/m3,是国家室内限值标准的12 倍。







 我要推广仪器
我要推广仪器
 下载APP
下载APP