[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=128099]相转移催化合成对羟基苯甲酸苄酯的研究[/url]可以借鉴里面的方法做研究的。
我从事表面活性剂的合成,请教对多羟基的季胺盐类物质的质谱,用什么方法作出分子离子峰比较有效?
接到一个含有N-羟基邻苯二甲酰亚胺的样品,有没有做过此分析的楼主,走捷径,请教N-羟基邻苯二甲酰亚胺在紫外吸收最佳波长是多少?
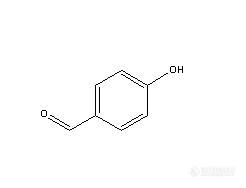
前几天,领导安排检测一个业务单位送来一个样品让我们帮忙调试方法,样品名称为对羟基苯甲醛,分子式见下图,样品为类白色粉末。http://ng1.17img.cn/bbsfiles/images/2010/05/201005242248_220486_1637960_3.jpg首先查找相关信息:对羟基苯甲醛熔点113-118℃,相对密度1.129(30/4℃)。广泛用于医药、香料、农药、石油化工、电镀等领域.在医药工业中,主要用合成羟氨苄青霉素(阿莫西林)、抗菌增效剂甲甲氧苄胺嘧啶(TMP)、3,4,5-三甲氧基甲醛、对羟基苯酐氨酸、羟氨苄头孢霉素、人造天麻、杜鹃素、艾司洛尔等;在香料工业中用于合成香兰素、乙基香兰素、洋茉莉醛、丁香醛、茴香醛和覆盆子酮等香料;在农药中主要用作除草剂溴苯腈和羟敌草腈的合成;在化工中主要用于合成对羟基苯甲酸、对羟基甲酸苄酯、醋酸对羟基苯酚酯 ;在国外还用于生产杀菌剂、照相乳化剂、镀镍光泽剂、液晶等。
多羟基化合物的H谱中,羟基的峰变宽了,有谁知道怎么处理啊?或者是怎么准备样品啊?谢谢啊
[color=#444444] 最近做了两个对比化合物的红外,一个是带有吗啉的萘酰亚胺,另一个是用盐酸将吗啉N成盐,用红外进行验证,,用KBr压片的方法做的红外,发现两个结构的红外都在3447位置有个较大的包峰,但是结构经核磁氢谱碳谱以及高分辨质谱的确认,结构中不含有羟基以及氨基等,只有一个吗啉环,一个萘酰亚胺、还有一个含硫的杂环。图谱分析不出该峰到底是什么基团的特征峰,请问有做红外的高手能指教一下吗?[/color]
3-羟基苯胺和3-氯苯胺缩合反应生成3,3'-二氨基二苯醚,反应具体的条件是什么???文献上只说了加催 化 剂Cucl,吡 啶 (Py),甲 苯和 碳 酸 钾,但没说加多少,所以请高人指点一下,十分感谢啦!!!!!http://emuch.net/bbs/images/smilies/smile.gifhttp://emuch.net/bbs/images/smilies/smile.gifhttp://emuch.net/bbs/images/smilies/smile.gifhttp://emuch.net/bbs/images/smilies/smile.gifhttp://emuch.net/bbs/images/smilies/smile.gif
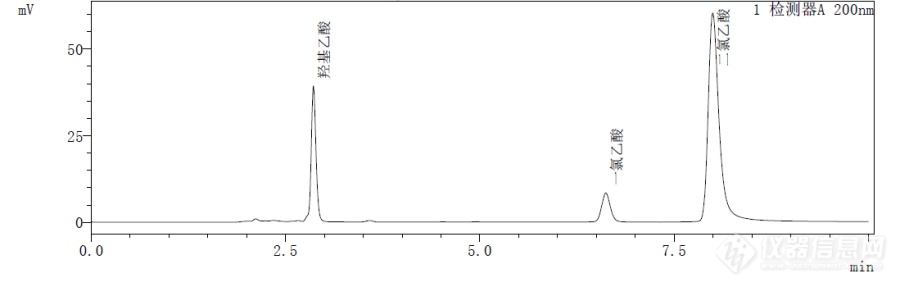
[align=center][b]椰油酰胺丙基甜菜碱中一氯乙酸、二氯乙酸和羟基乙酸的测定[/b][/align] 椰油酰胺丙基甜菜碱(CAB)是一种两性表面活性剂,因其对眼睛和皮肤刺激性低,对头发和皮肤有护理效果并产生大量稳定泡沫,在肥皂和硬水中有出色的起泡性和洗涤性,故广泛用于香波和泡沫浴液等洗涤用品中。 在工业生产中,常使用一氯乙酸(MCA)作为原料生产CAB。而工业MCA中含有少量的二氯乙酸(DCA),DCA是生物学证实具有潜在致癌风险的物质,同时在生产过程中残留的MCA对皮肤、黏膜有很强的腐蚀性,通常采用水解法将MCA转化为刺激性更小的羟基乙酸(GCA)。椰油酰胺丙基甜菜碱产品的指标含量分析中,一般要求一氯乙酸<20ppm,二氯乙酸<300ppm,羟基乙酸<0.5%。[b]色谱条件:[/b]色谱柱:[b]Kromasil C8(4.6*250mm,5μm)[/b]柱 温:24℃检测器:紫外检测器波 长:200nm流动相:乙腈:水=10:90(每1000mL中加入2.0mL磷酸)流 速:1ml/min进样体积:20μL采集时间:10min[img=,690,219]https://ng1.17img.cn/bbsfiles/images/2018/10/201810291003374445_9066_2428063_3.png!w690x219.jpg[/img] 图1 :一氯乙酸、二氯乙酸和羟基乙酸混标色谱图[img=,690,328]https://ng1.17img.cn/bbsfiles/images/2018/10/201810291003547039_780_2428063_3.png!w690x328.jpg[/img] 图2 :椰油酰胺丙基甜菜碱样品色谱图[b]总结[/b]参考国标GB/T 28193-2011表面活性剂中氯乙酸(盐)残留量的测定方法,建立高效液相色谱法,一次性测定样品中一氯乙酸、二氯乙酸和羟基乙酸的含量。其优点是以高比例水相作为流动相,样品不需要进行萃取、酯化等前处理,操作方便,快速高效。使用Kromasil C8色谱柱分离样品中一氯乙酸与其余组分,效率高,分离度好,结果可靠,可为椰油酰胺丙基甜菜碱生产厂家提高产品质量提供参考。[b]注:由深圳爱湾医学检验实验室验证 [/b]
如题。希望能找到苄胺的标准,国标、行标、国外标准都可。谢谢。
两种单体共聚合,丙烯酰胺和丙烯酸,两者只是一个氨基和羟基的区别,如果用氘代水做氢谱,是不是羟基峰氨基峰会与溶剂峰重叠? 即使不重叠,似乎也氨基羟基化学位移也相似? 那如果用碳谱呢?因为共聚物是水溶性的,在溶剂方面除了氘代水似乎没有其他的选择了.先谢谢大家了! 另祝大家情人节快乐!
各位大神好,新人小白一枚,想请教一下利用安捷伦GC 7890B测苄胺和其氧化产物。我用的乙酸乙酯做溶剂,试过HP-5的色谱柱,没出峰,然后现在用HP-35,也加大了苄胺浓度,在六分钟出峰了,峰很奇怪,不知道是拖尾峰严重还是产生其他物质没分离?怎么解决这个问题?麻烦各位大神指教!
各位大神好,新人小白一枚,想请教一下利用安捷伦GC 7890B测苄胺和其氧化产物。我用的乙酸乙酯做溶剂,试过HP-5的色谱柱,没出峰,然后现在用HP-35,也加大了苄胺浓度,在六分钟出峰了,峰很奇怪,不知道是拖尾峰严重还是产生其他物质没分离?怎么解决这个问题?麻烦各位大神指教!
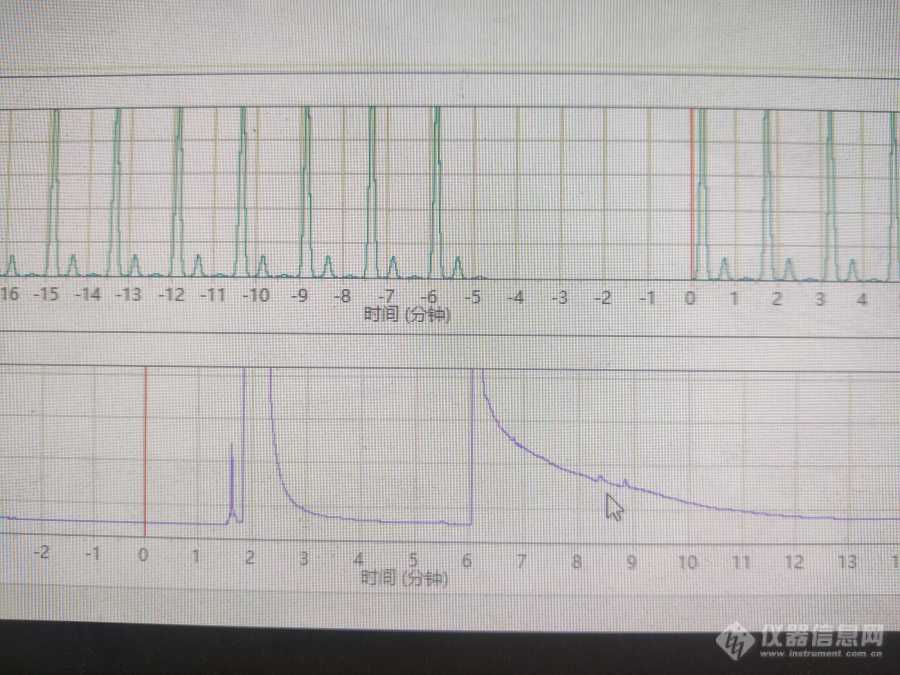
各位大神好,新人小白一枚,想请教一下利用安捷伦GC 7890B测苄胺和其氧化产物。我用的乙酸乙酯做溶剂,试过HP-5的色谱柱,没出峰,然后现在用HP-35,也加大了苄胺浓度,在六分钟出峰了,峰很奇怪,不知道是拖尾峰严重还是产生其他物质没分离?怎么解决这个问题?麻烦各位大神指教![img]https://ng1.17img.cn/bbsfiles/images/2023/04/202304291622315276_421_5986350_3.jpg[/img]
如题;某个生产过程使用苄醇和另一个化合物A反应生产磺酰胺;可是磺酰胺紫外响应很弱,因此想尝试CAD检测器;但是CAD上没有找到苄醇和磺酰胺的峰,是不适合用这个检测器么?
各位大神好,新人小白一枚,想请教一下利用安捷伦GC 7890B测苄胺和其氧化产物。我用的乙酸乙酯做溶剂,试过HP-5的色谱柱,没出峰,然后现在用HP-35,也加大了苄胺浓度,在六分钟出峰了,峰很奇怪,不知道是拖尾峰严重还是产生其他物质没分离?怎么解决这个问题?麻烦各位大神指教![img]https://ng1.17img.cn/bbsfiles/images/2023/04/202304291630013379_13_5986350_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/04/202304291630014088_4008_5986350_3.png[/img]
想要做 苄索氯胺 的 液相色谱检测哪里可以购买到 苄索氯胺 的标准样品拜托了留言或邮箱 linsheng@fzyamazaki.com谢谢
最近做的一个化合物,配体上有两个羟基,但是核磁显示的是四个羟基峰,测了二维核磁和变温核磁,仍然不能解释,求高手指点。谢谢
【序号】:1【作者】: 张雅伦【题名】:聚乙二醇的N-羟基琥珀酰亚胺琥珀酸酯的合成工艺、苏氨酸负载及催化应用【期刊】:兰州大学【年、卷、期、起止页码】:2017【全文链接】:https://kns.cnki.net/kcms2/article/abstract?v=3uoqIhG8C475KOm_zrgu4lQARvep2SAkOTSE1G1uB0_um8HHdEYmZhkBIZJEK02VaOdneXeYijuWwpOfpIhlJTd0mjIpAyz7&uniplatform=NZKPT
[color=#444444]最近做苄胺偶联反应,乙腈作溶剂,分析反应结果时,发现个问题,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]没有峰,色谱柱是弱极性柱。环己烷+苄胺,正常出峰;乙腈+苄胺,没有明显的峰,只有些小峰,怀疑是杂质。如果说是因为柱子是弱极性,而乙腈是极性的问题,可是乙醇却能正常出峰。。。求大神分析,感激不尽[/color]
《CNAS T0441食品(果蔬汁)中羟基苯甲酸酯、环己基胺基磺酸纳、日落黄和酒石黄的检测》能力验证开始了,大家保持联络啊~~呵呵~
哪位同志有对羟基苯乙酮的滴定含量检测方法.我这里有一个方法,但总是滴不好,用甲醇钠溶液进行滴定,用二甲基甲酰胺进行溶解,用麝香草酚蓝作指示剂.目前存在的问题就是终点变化不明显.哪位有用到或碰到类似情况,都来说说吧.
如何用液相色谱分析带羟基和氨基的磺酰胺类染料中间体和染料,用什么柱子、什么流动相?谢谢专家
[color=#444444]有人知道苄胺做溶剂残留时,[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测方法及色谱条件吗?谢谢啦[/color]
增效联磺片为磺胺类抗菌消炎药的新型复方制剂,每片含磺胺甲基异(口恶)唑200mg、磺胺嘧啶200 mg、甲氧苄氨嘧啶80 mg,各地方标准均有收载,对前两种成分以纸色谱法鉴别,而对甲氧苄氨嘧啶则另行鉴别。本文以薄层色谱法同时鉴别磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶3种成分,专一性强,斑点明显,操作简便,结果较为满意。1 仪器与试药 三用紫外线分析仪(上海顾村电光仪器厂),硅胶GF254薄层板(10cm×20 cm,自制);磺胺甲基异(口恶)唑、磺胺嘧啶和甲氧苄氨嘧啶对照品(中国药品生物制品检定所);增效联磺片(市售品);硅胶GF254(青岛海洋化工厂生产,化学纯);其它试剂均为分析纯。2 溶液的配制2.1 单一对照品溶液 分别精密称取磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶对照品适量,加50%丙酮溶液分别制成0.5 mg/mL磺胺甲基异(口恶)唑、0.5 mg/mL磺胺嘧啶、0.2 mg/mL甲氧苄氨嘧啶的单一对照品溶液。2.2 混合对照品溶液 精密称取磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶对照品适量,加50%丙酮溶液制成? mL含磺胺甲基异(口恶)唑0.5 mg、磺胺嘧啶0.5 mg和甲氧苄氨嘧啶0.2mg的混合对照品溶液。2.3 样品溶液的配制 取供试品细粉适量(约相当于磺胺甲基异(口恶)唑50mg),加50 %丙酮溶液100 mL,振摇使溶解,过滤,滤液作为供试品溶液。
欧盟委员会发布官方公报(EU)No358/2014,修订了欧洲化妆品法规No1223/2009附件Ⅱ,限制物质清单新增尼泊金异丙酯、羟苯异丁酯、羟苯苄酯、4-羟基苯甲酸苯酯、戊烷基对羟苯甲酸酯5种对羟基苯甲酸酯类物质。 此外,修订案还规定二氯苯氧氯酚在漱口水中使用最大浓度为0.2%,在其他化妆品如牙膏、手皂、扑面粉中使用最大浓度为0.3%。羟基苯甲酸及其盐和酯类作为单酯中的酸用于制作配制品中的最大浓度为0.4%,作为混合酯中的酸最大允许浓度为0.8%。2014年10月30日前,不符合新规的化妆品仍可在市场上正常销售,2015年6月30日起,所有市场上流通的化妆品必须符合新规。 对此,检验检疫部门提醒相关企业:一是密切关注欧盟化妆品修订案,及时掌握法规变化动态;二是强化同进口商的沟通,做好过渡期期间的合同评审,避免因法规认识偏差导致的退运风险;三是加强产品质量管控,通过优化升级生产工艺、第三方检测,确保降低对羟基苯甲酸酯类限制物质含量,确保平稳过渡。
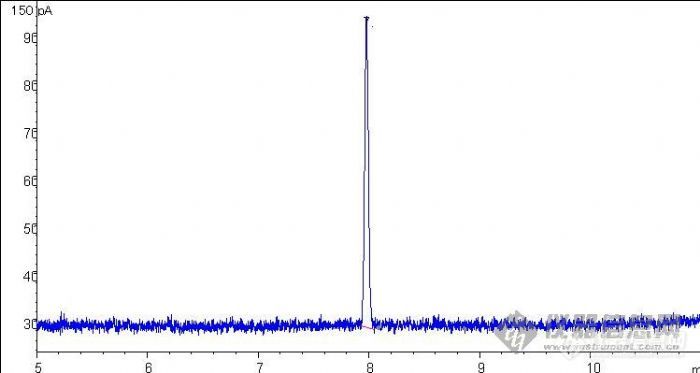
前段时间做甲胺磷的检测,一直做的挺好的,而且峰型也漂亮,但是从昨天开始,峰变的展宽很矮,我什么条件也没有变,柱子刚开始用有半个月,就算每天平均打20个样,一共才没有多少啊,况且这个柱子只做甲胺磷用,其他什么都没有做过。我做过老化,最高温度250度烧了半个小时,也换了隔垫和衬管了,其实这都才没有用多久,我用的7890是新仪器。但是,这些做后仍然是没有改变,请教大家,一下是图谱:[img]http://ng1.17img.cn/bbsfiles/images/2008/06/200806110944_92560_1614502_3.jpg[/img]原来100ppb甲胺磷[img]http://ng1.17img.cn/bbsfiles/images/2008/06/200806110944_92561_1614502_3.jpg[/img]现在100ppb甲胺磷
8-羟基喹啉重量法(GB/T 5059.1-1985)1.方法提要试样以硝酸-氯酸钾饱和溶解分解,钼成钼酸析出,用EDTA络合铁等杂质,加氨水溶解钼酸按,过滤。取部分溶液,pH约4.8醋酸-醋酸按缓冲溶液中,以8-羟基喹啉沉淀钼用玻璃坩埚过滤,在130~140℃电热干燥箱内干燥称重,借此测得钼的含量。本方法适用于钼精矿中钼量的测定。测定范围:大于10%2.试剂氨水(比重0.9)硝酸-氯酸钾饱和溶液:加氯酸钾在硝酸(比重1.42)中,成饱和溶液。乙二胺四乙酸二钠ETDA(C10H12FeN2NaO8• 3H2O)(5 %)。甲基橙(0.1%)。盐酸(1+1)。醋酸-醋酸铵缓冲溶液(pH4.8):280克醋酸铵,用水溶解后,加220毫升冰醋酸,再用水稀释至1000毫升,混匀。8-羟基喹啉(3%):30克8-羟基喹啉溶解在1000毫升2mol/L醋酸中(冰醋酸118毫升)。3.分析步骤称取0.2305克试样,置于250毫升烧杯中,加15~20毫升硝酸-氯酸钾饱和溶液,待剧烈作用后,低温加热分解并蒸发至溶液约剩5毫升,取下,冷却。用少量水吹洗表面皿及烧杯内壁,加20毫升5 % ETDA溶液,摇匀,用氨水(比重0.9)中和至钼酸沉淀全部溶解并过量2毫升,冷却至室温,移入100毫升容量瓶中,用慢速滤纸干过滤,移取50.00毫升试液,置于250毫升烧杯中,加1滴0.1%甲基橙指示剂,用盐酸(1+1)中和至溶液颜色刚好变红,加5毫升醋酸-醋酸按缓冲溶液,用水稀释至溶液约120毫升,煮沸,取下。在不断搅拌下,徐徐加10毫升3%8-羟基喹啉溶液,置于低温电炉上静置2~3分钟。取下,用已在130~140℃干燥过并称重的3~4#玻璃坩埚过滤,用热水洗涤烧杯3~4次,洗涤沉淀8~10次,将坩埚连同沉淀置于130~140℃电电热干燥箱中干燥1小时,取出,置于干燥器内冷却至室温,称重。并反复千燥至恒重。钼的百分含量按下式计算; 式中;W1——玻璃坩埚与8-羟基喹啉钼的重量(克);,W2——玻璃坩埚的重量(克);V——试液总体积(毫升);V1——分取试液体积(毫升);W——称样量(克);0.2305——8-羟基喹啉钼换算成钼的系数。4.允许差含钼量(%)允许差(%)≤200.2520.01~3000.3030.000.405.注意事项①加硝酸-氯酸钾饱和溶液时应防止加入固体氯酸钾。如试样分解不完全应重复加硝酸-氯酸钾和溶液至试样分解完全。②此时不宜将溶液煮沸,否则沉淀会溅跳粘附在烧杯内壁上不易洗净。
求助,香草扁桃酸和3-甲氧基-3-羟基苯乙二醇衍生物GC-MS分不开,不管是特征离子还是保留时间都相近怎么办,RT,升温程序改善了没用,柱子是DB-5
[size=3][b]有没谁有4-甲基-5,7-二羟基香豆素的1H谱图[/b][/size]我们学校核磁仪器坏了,答辩前都修不好,希望有此化合物的图的可以给我一张,最好是扫描的。。。。谢谢了。
有做过8-羟基喹啉或8-羟基喹啉硫酸盐的液相实验吗?为什么分不开啊,用的是化妆品检疫的方法,求助!求助!







 我要推广仪器
我要推广仪器
 下载APP
下载APP