循环伏安法 制得的高分子薄膜侧基咔唑作为交联点生成二聚咔唑,想测其反应的咔唑基团数占起始的咔唑基团数的比值谢谢大家,用红外的方法可以么?怎么用?O(∩_∩)O谢谢
谁知道哪里卖知道确切气相色谱含量的咔唑样品啊,只要含量达到九十以上就行啊,知道的朋友请回答啊,谢谢啊。
(一)食品中果胶的意义果胶产品可分为高脂果胶和低脂果胶,酯化度大于50%的称为高脂果胶,酯化度低于50%的称为低脂果胶,果胶的显著特性是有胶凝性和增稠稳定性,并且有很强的耐酸、耐高温性能。果胶在食品工业中应用较广,如利用果胶水溶液在适当条件下可以形成凝胶的特性,生产果酱、果冻及高级糖果等食品;利用果胶具有增稠、稳定、乳化等功能,可以解决饮料的分层、防止沉淀的问题,还可以改善风味等。测定果胶物质的方法有称量法、咔唑比色法、果胶酸钙滴定法、蒸馏滴定法等。(二)咔唑比色法果胶经水解可生成半乳糖醛酸,半乳糖醛酸在强酸中可与咔唑试剂发生缩合反应,生成紫红色化合物,该紫红色化合物的呈色强度与半乳糖醛酸含量成正比,故可通过测定吸光值对果胶含量进行定量。此法适用于各类食品的果胶含量的测定,具有操作简便、快速、准确度高、重现性好等特点。1.样品处理同重量法。2.果胶处理同重量法。3.标准曲线制作取8支50mL比色管,各加入12mL浓硫酸,于冰水浴中边冷却边缓缓依次加入浓度为0、10μg/mL、20μg/mL、30μ/ml、40μ/mL、50μg/mL、60μg/mL、70μg/ml的半乳糖醛酸标准溶液2mL,充分混合后,再置于冰水浴中冷却。然后在沸水浴中准确加热10min,迅速冷却到室温,各加入0.15%咔唑试剂1mL。充分混合,室温下放置30min,以半乳糖醛酸含量为0的半乳糖醛酸标准溶液为空白,在530nm波长下测定吸光值,以半乳糖醛酸含量为纵坐标,吸光值为横坐标,绘制标准曲线。4.样品提取液的测定取果胶提取液,用水稀释到适当浓度(含半乳糖醛酸10~70μg/mL)。取2mL稀释液于50mL比色管中,以下按制作标准曲线的方法操作,测定吸光值。从标准曲线上查出半乳糖醛酸的浓度(μg/mL)。5.结果计算X(以半乳糖醛酸计)=c*V*K/m*106*100式中 X——样品中果胶的质量分数,%;c——从标准曲线上查得的半乳糖醛酸的浓度,肚g/mL;V——果胶提取液的总体积,mL;K——提取液稀释倍数;m——样品质量,g。6.试剂①乙醇。②乙醚。③0.05mol/L盐酸溶液。④O.15%咔唑乙醇溶液:化学纯咔唑0.150g,溶解于精制乙醇中并定容到100mL,咔唑溶解缓慢,需加以搅拌。精制乙醇:取无水乙醇或95%乙醇1000mL,加入锌粉4g,(1+1)硫酸4mL,在水浴中回流10h,用全玻璃仪器蒸馏,馏出液每1000mL加锌粉和氢氧化钾各4g,重新蒸馏一次。⑤半乳糖醛酸标准溶液:半乳糖醛酸100mg,溶于蒸馏水并定容到100mL,用此液配制一组浓度为10~70μg/mL的半乳糖醛酸标准溶液。⑥硫酸。7.仪器①分光光度计。②经50mL比色管。8.注意事项①本法的测定结果以半乳糖醛酸表示,不同来源的果胶中半乳糖醛酸的含量不同,如甜橙为77.7%,柠檬为94.2%,柑橘为96%,苹果为72%~75%,若把结果换算为果胶的含量,可按上述关系计算换算系数。②样品处理时应充分洗涤去除糖分,减少其存在对咔唑的呈色反应的影响。③在测定样液和制作样液标准曲线时,应使用相同规格、同批号的浓硫酸,以保证浓度一致,减少硫酸浓度对咔唑的呈色反应的影响。
【关键词】资料分享 可以有 做广告 不许有 ——土豆批改 内容摘要:果胶经水解生成半乳糖醛酸,在硫酸中与咔唑试剂发生缩合反应,生成紫红色化合物,其呈色强度与半乳糖醛酸含量成正比,可比色定量。 (中检所标准物质) 咔唑比色法 此法适用于各类食品,且操作简便、快速,准确度高. 1.原理 果胶经水解生成半乳糖醛酸,在硫酸中与咔唑试剂发生缩合反应,生成紫红色化合物,其呈色强度与半乳糖醛酸含量成正比,可比色定量。 2.仪器 ①分光光度计。 ②50 mI,比色管。 3.试剂 ①99%乙醇。 ②70%乙醇. ③yi醚。 ④0.05 tool·L叫盐酸溶液。 ⑤0.5。tool·L叫氢氧化钠。 ⑥O.15%咔唑乙醇溶液:称取化学纯咔唑O.15 g,溶解于精制乙醇中并定容到1。0 mI咔唑溶解缓慢,需加以搅拌。 ⑦精制乙醇:取无水乙酵或95%乙醇1 000 mL,加入锌粉4 g,硫酸(1:1)4 mI_,,在水浴中回流10 h,用全玻璃仪器蒸馏,馏出液每1 000 mI.加锌粉和氢氧化钾各4 g,重新蒸馏一次。(药检所对照品) ⑧半乳糖醛酸标准贮备溶液:准确称取半乳糖醛酸100 mg,溶于蒸馏水并定容到100mL,得浓度为l mg·mI.1半乳糖醛酸标准贮备液。 ⑨半乳糖醛酸标准工作液:分别准确吸取0.0、1.0、2.O、3.O、4.O、5.O、6.O、7.0 ml。半乳糖醛酸标准贮备溶液于8个10¨。mL容量瓶中,用水稀释至刻度,得一组浓度分别为0.0、10、20、30、40、50、60、70扯g·mI。叫的半乳糖醛酸标准工作液。 ⑩浓硫酸:优级纯。 4.测定步骤 ①提取果胶 a.水溶性果胶提取:用150 mI,水将上述挥发至干的残渣移人250 mI.烧杯中,加热至沸并保持沸腾1 h,随时补足蒸发的水分,冷却后移人250 mI。容量瓶中,加水定容,摇匀,过滤,弃去初滤液,收集滤液即为水溶性果胶提取液。 b.总果胶的提取:用150 mL加热至沸的0.05 tool·L叫盐酸溶液把挥干的残渣移人250mL锥形瓶中,装上冷凝器,于沸水浴中加热回流1 h,冷却后移人250 ml。容量瓶中,加甲基红指示剂2滴,加O.5 mol·L1氢氧化钠中和后,用水定容,摇匀,过滤,收集滤液即为总果胶提取液。(北京标物中心) ②样品处理 a.新鲜样品:称取试样30~50 g,用小刀切成薄片,置于预先放有99%乙醇的500 mI。锥形瓶中。装上回流冷凝器,在水浴上沸腾回流15 rain后,冷却。用布氏漏斗过滤,残渣移至研钵中,一边慢慢磨碎,一边滴加70%的热乙醇,冷却后再过滤,反复操作至滤液不呈糖的反应(用苯酚一硫酸法检验,见说明)为止。残渣用99%乙醇洗涤脱水,再用yi醚洗涤以除去脂类和色素,漏斗中残渣在空气中挥发去yi醚。 b.干燥样品:研细,过60目筛,称取5~10 g样品于烧杯中,加入热的70%乙醇,充分搅拌以提取糖类,过滤。反复操作至滤液不呈糖的反应。残渣用99%乙醇洗涤,再用yi醚洗涤,最后让yi醚挥发掉。 ③标准工作曲线的制作:取8支50 mL比色管,各加入12 mI。浓硫酸,置冰水浴中,边冷却边缓缓依次加入浓度为O.0、10、20、30、40、50、60、70肚g·ml,叫的半乳糖醛酸标准溶液2mL,充分混合后,再置冰水浴中冷却。然后在沸水浴中准确加热10 min,用流动水迅速冷却到室温,各加人O.15%咔唑试剂1 mI。,充分混合,置室温下放置30 rain,以O号管为空白在530nm波长下测定吸光度,绘制标准工作曲线。(中华标准物质网) ④测定:取果胶提取液,用水稀释到适当浓度(含半乳糖醛酸10~70肛g·mLl)。取2ml。稀释液于50 mI.比色管中,以下按制作标准曲线的方法操作,测定吸光度。从标准曲线上查出半乳糖醛酸浓度(ug·mI。) 5.计算 6.说明 ①糖分的存在对咔唑的呈色反应影响较大,使结果偏高,故样品处理时应充分洗涤以除去糖分。 ②检验糖分的苯酚一硫酸法:取检液l mL,置于试管中,加入5%苯酚水溶液1 ml。,再加入硫酸5 mL,混匀,如溶液呈褐色,证明检液中含有糖分。 ③硫酸浓度对呈色反应影响较大,故在测定样液和制作标准曲线时,应使用同规格、同批号的浓硫酸,以保证其浓度一致。土豆:感谢分享资料,但是含有广告内容是不允许的哦,亲,下次别违规了。
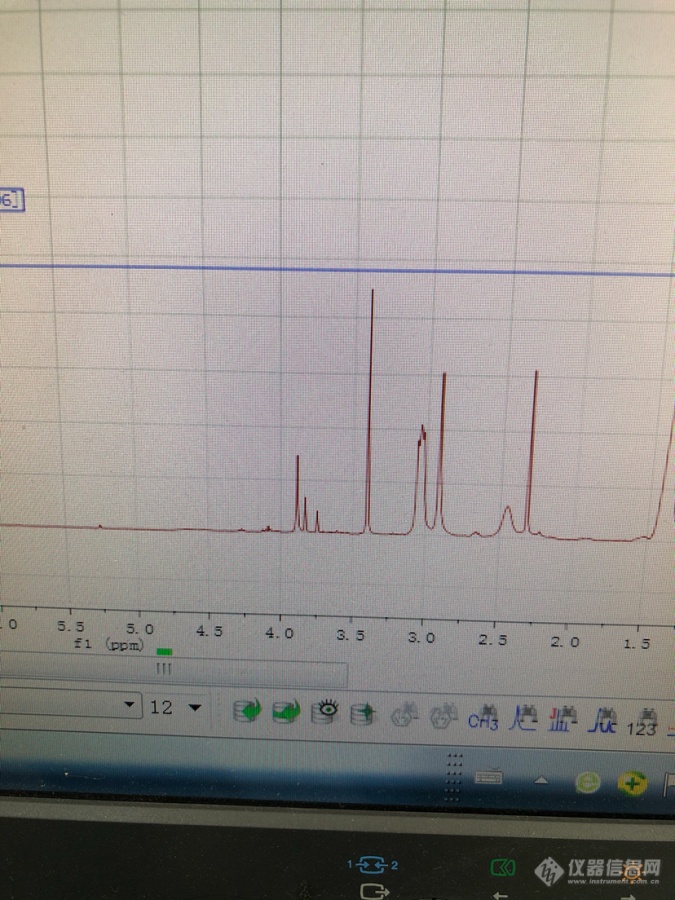
[color=#444444]做的是聚(2,7-咔唑)衍生物 [/color][color=#444444]甲苯作溶剂 反应后萃取浓缩[/color][color=#444444]可是滴入甲醇中 无沉淀 最后都是粘稠的液体 无固体沉出[/color][color=#444444]所以反映结束后处理该怎么做呢 多次进行溶解沉出 都是沉不出固体[/color][color=#444444]测核磁3-4的峰 可能是甲苯和甲醇未干吗?[/color][color=#444444][img=,675,900]https://ng1.17img.cn/bbsfiles/images/2019/05/201905151636301368_4040_1646718_3.png!w675x900.jpg[/img][/color]
[color=#444444]请问各位大侠,羧酸和羧酸盐在同一液相色谱条件下出峰的时间是否一样?例如醋酸和醋酸钠,流动相为乙腈和水,PH为酸性,磷酸做缓冲液。另外,羧酸和羧酸盐的极性是否相同?[/color]
请问同志们,羧酸类物质能检测吗??能用甲醇做流动相吗?羧酸会不会和甲醇生成酯,而影响测定羧酸的PH=3-4,需要配制流动相+怎么样的缓冲溶液??????大家帮忙啊
求用羧酸合成酰 氯,羧酸分子350以上,酰 氯如何分析,酰 氯反应如何中控?
HJ 1220-2021 6种羧酸类的测定 [url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]质谱法 一般的柱子的可以分析吗
我看的文献方法衍生全氟羧酸,用三乙基硅烷醇的方法,用的仪器是岛津的单杆EI 源,但是衍生以后全扫模式下,所有的全氟羧酸出的峰都一样。通过SIM模式下才能找到目标峰,并且PFDA/PFNA/PFDOA的峰都非常小。我用的是1ug/ml得标液衍生的,全氟辛酸的峰大概只有1000,其他的峰高就只有100不到。有没有大神做过类似的方面,求帮助。还有一个问题,如果做全氟羧酸的目标物,用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]做的话,文献中有用NCI源和EI源的,具体的那个方法更好一点呢。跪谢!
工作场所羧酸类,我只有ffap极性毛细管柱,方法中乙酸是用甲酸作为溶剂,可是甲酸腐蚀性太大,不敢做,有没有其它溶剂能代替的,还有氯乙酸响应实在太小,峰型很难看,检出限远达不到国标,是色谱柱的问题吗?哪位同行做过,详解一下
请问:羧酸类打[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]峰型总是不好,怎么办?
做金属物质检测时,会用的吡啶-2,6-二羧酸,皮考啉二酸,DPA,这个物质,不知道有没有那个老师以前做过?实验过程中,起到什么作用呢?谢谢!
羧酸键合硅胶柱用前需要怎么处理,用后如何保养,求指教
买了一根羧酸键合硅胶柱要保存在叠氮钠里边,但采购说没有优级纯只有分析纯,可以吗
吡啶二羧酸酐结构看似简单,可因其在水中水解成酸,影响样品游离酸含量分析,化学滴定不好作;做GC时热分解,与相应的二酸峰重合;液相用水溶液作流动相不行,用非水有机相作流动相如用醇又会醇解,估计可用正相做,但该样在许多有机溶剂中溶解性差,所以一直没找到好的分析方法。请问哪位老师作过吡啶二羧酸酐的HPLC分析,能否帮帮我。谢放!
[font=Verdana]羧酸吸收什么情况下会消失?[/font]除了重水交换以外,还有别的情况会导致羧酸上的H的核磁吸收消失吗?
如何利用红外确认羧酸根与金属离子的配位形式?
减水剂是一种重要的混凝土外加剂,能够最大限度地降低混凝土水灰比,提高混凝土的强度和耐久性。减水剂分为普通减水剂和高效减水剂,减水率大于5%小于10%的减水剂称为普通减水剂,如松香酸钠、木质素磺酸钠和硬脂酸皂等;减水率大于10%的减水剂称为高效减水剂,如三聚氰胺系、萘系、氨基磺酸系、改性木质素磺酸系和聚羧酸系等。在众多高效减水剂中,具有梳形分子结构的聚羧酸系高效减水剂因其减水率高、坍落度保持性能良好、掺量低、不引起明显缓凝等优异性能,成为近年来国内外研究和开发的重点。 在国外,聚羧酸类减水剂的研究已有相当长的历史,其应用技术已经成熟。日本是研究和使用聚羧酸类减水剂最多也是最成功的国家,1995年以后聚羧酸系减水剂在日本的使用量就超过了传统的萘系减水剂,1998年底聚羧酸系减水剂产品已占所有高性能AE减水剂产品总数的60%以上,其主要生产厂商有花王、竹本油脂、日本制纸、藤泽药品等[1]。对聚羧酸系减水剂的研究主要集中在新拌混凝土有关性能和硬化混凝土的力学性能及高强高性能混凝土在工程中的应用技术。目前聚羧酸系减水剂可使混凝土的水灰比下降到0.25以下,而水泥用量仍可保持在500 kg/m3,同时它的坍落度可保持200 mm以上,完全满足施工要求。近年来,北美和欧洲的一些研究者的论文中也有许多关于研究开发具有优越性能的聚羧酸系减水剂的报道,主要是商业开发和推广,如Grance公司的Adva系列、MBT公司的pheomixTOOFC牌号、Sika公司的Viscocrete3010等。 由于成本和技术性能问题,国内对聚羧酸类减水剂产品的研究仅处于实验室研制阶段,只有少量用作坍落度损失控制剂与萘系减水剂复合使用。而且可供合成聚羧酸类减水剂的原料也极为有限,国内原材料单甲氧基聚乙二醇(MPEG)供应不足,MPEG国内没有商业化,必须依靠进口,也有研究人员[3]用聚乙二醇(PEG)代替MPEG,但是由于在制备过程中,双官能度的PEG容易产生交联,使得产品性能较差,质量不稳定。可以说,从减水剂原料到生产工艺、降低成本、提高性能等许多方面都仅仅是处于刚起步阶段。 [color=#DC143C]本文主要对聚羧酸系高效减水剂的化学结构、主要作用机理、合成方法及结构与性能的关系进行了综述。[/color]
要测葡萄糖中C2-C5的一元羧酸检测方法请高手执教!
我想做三羧酸循环中a-酮戊二酸标记或乙酰辅酶A中碳的同位素标记然后研究三羧酸循环和这种植物所形成的色素的关系,可是我没有同位素标记设计的经验,谁可以帮帮我,谢谢啦~~~
最近做的样品中含有一些羧酸还有醛类,看了一些文献,对于醛类,有的用DNPH(2,4-二硝基苯肼),有的用PFBHA。对于羧酸,有的用BSTFA。请问1、应当如何对样品进行衍生化呢?2、衍生化试剂一般从哪里买的?看了一些国外的试剂公司,衍生化试剂非常贵,动辄就上千元,不敢乱来。谢谢各位!
买了一根羧酸键合硅胶柱要保存在叠氮钠里边,但采购说没有优级纯只有分析纯,可以吗
1.O-H伸缩振动羧酸在浓溶液或固态中因强氢键成二聚体。 强氢键源于离子共振,阻碍游离羟基振动,仅稀非极性溶剂或蒸气相中可见(约3520 cm?1)。 二聚体O-H振动宽且强,范围3300~2500 cm?1,常集中于3000 cm?1,伴弱C-H振动。 长波长精细结构为倍频与复合频。 β-二酮等也有此吸收,但较弱,C-O振动频率较低。与醚类溶剂形成分子间氢键,O-H吸收约3100 cm?1。 2.C-O伸缩振动羧酸C-O振动强于酮,单体约1760 cm?1。二聚体对称,仅不对称振动有吸收,氢键与共振降低频率至1720~1706 cm?1。 分子内氢键影响更大,如水杨酸1665 cm?1,对羟基苯甲酸1680 cm?1。 不饱和共轭轻微降低频率,α,β-不饱和及芳基共轭酸二聚体约1710~1680 cm?1。 α位电负性取代基(如卤素)轻微增加频率,旋转异构致双重谱带。 3.C-O伸缩与O-H弯曲振动羧酸红外光谱中,C-O伸缩约13201210 cm?1,O-H弯曲约14401395 cm?1,两者有相互作用。 二聚体C-O伸缩强吸收约1315~1280 cm?1,长链脂肪酸呈双峰。 O-H面外弯曲特征谱带约920 cm?1,中等强度峰宽。
我想做放射性同位素标记三羧酸循环中间产物,14C标记a-酮戊二酸,柠檬酸和琥珀酰辅酶A,可是这方面的资料我找到很少,希望你们能帮帮我啦~
一个实例,用七氟丁酸酐可以与二苯基甲烷二胺发生衍生化反应,衍生化的机理应该在于胺根上面。但二苯基甲烷二胺里面的胺还会与羧酸发生反应。如果同时向含有 大量的七氟丁酸酐 和 极少量的 丁酸 的混合溶剂中,加入少量二苯基甲烷二胺,请问二苯基甲烷二胺是先发生衍生化反应还是与丁酸反应?
有没有做过喹那啶-4羧酸的液相分析的伙伴们?
羧酸的活泼氢在氢谱里不愿出峰的原因??
如何能使羧酸上羟基的氢原子显示出来?谢谢!
1.C…O键强:介于C=O与C―O之间,强偶合。 2.光谱特征 不对称伸缩谱带:1650~1550 cm?1,显著。 对称伸缩谱带:1400 cm?1附近,较弱。 3.结构确证方法 转化为盐:羧酸与脂肪族叔胺(如三乙胺)在氯仿中反应(四氯化碳无效)。 4.特征谱带 羧酸根离子:两个羰基吸收谱带。 “铵”谱带:2700~2200 cm?1。 O—H伸缩谱带:消失。



 我要推广仪器
我要推广仪器
 下载APP
下载APP