偏钒酸铵 纯度怎么测定 采用什么方法 , 怎么样证明测定的是偏钒酸铵 而不是五价钒
请问:偏钒酸铵在水中的溶解度是多少?
请问专家:用什么仪器或仪器组合可以分析含六偏磷酸钠,偏矾酸铵,偏矾酸钠,硫酸钠,硼砂这样的溶液(各组分含量在2%左右)?
[b]1. 实验方法1.1 仪器与试剂1.1.1 仪器 [/b] 仪器为美国PerkinElmer Optima 7300 DV全谱直读电感耦合等离子体原子发射光谱仪,测试波长范围169~780nm,双向观测技术,在等离子体气、辅助气和雾化气气路上采用质量流量计,采用40.68MHz自激式射频发生器,高性能二维交叉色散中阶梯光栅分光系统,双专利设计的SCD固态检测器,正交雾化器。美国Milli-Q纯水器。仪器测定钒时所采用的工作参数列于表1。[b] 表1 仪器工作参数[/b] [table=596][tr][td] [align=center][b]波长[/b][/align] [align=center][b](nm)[/b][/align] [/td][td] [align=center][b]发射功率(W)[/b][/align] [/td][td] [align=center][b]雾化气流速[/b][/align] [align=center][b](L[b]×[/b]min[sup]-1[/sup])[/b][/align] [/td][td] [align=center][b]等离子体气流速[/b][/align] [align=center][b](L×min[sup]-1[/sup])[/b][/align] [/td][td] [align=center][b]辅助气流速[/b][/align] [align=center][b](L×min[sup]-1[/sup])[/b][/align] [/td][td] [align=center][b]样品提升速度[/b][/align] [align=center][b](mL×min[sup]-1[/sup])[/b][/align] [/td][/tr][tr][td] [align=center]290.880[/align] [/td][td] [align=center]1300[/align] [/td][td] [align=center]0.8[/align] [/td][td] [align=center]15[/align] [/td][td] [align=center]0.2[/align] [/td][td] [align=center]1.5[/align] [/td][/tr][/table][b]1.1.2 试剂[/b] GBW(E)080216钒单元素标准溶液(r=100 ug/mL,中国计量科学研究院,北京),实验中使用的标准工作溶液是通过用4%盐酸逐渐稀释而获得的;HNO[sub]3[/sub]、HCl(MOS级试剂,天津市风船化学试剂科技有限公司);所用水为经过Milli-Q纯水器(Millipore公司,美国)纯化后获得的电阻率为18.2 MW×cm的超纯水。[b]1.2 实验方法1.2.1 建立的银铜钒合金样品前处理方法[/b] 准确称取0.1000 g左右的银铜钒合金样品到玻璃烧杯中,分别加入5 mL的MOS级浓硝酸,加盖置于电热板上低温加热消解,待样品溶液变清亮后,升温将过剩的HNO[sub]3[/sub]蒸至小体积,取下、冷却至室温,定容到25 mL容量瓶中,待测。并同时作样品空白实验。[b]1.2.2国家标准中的样品前处理方法[/b] 根据GB/T 15072.19- 2008标准方法[sup][/sup],样品的前处理过程如下:将试料置于150 mL烧杯中,加入2 mL硝酸,盖上表面皿。低温加热至试料完全溶解。再加入20 mL水稀释,加入2mL盐酸,搅拌,微沸20 min至氯化银完全沉淀凝结。用中速滤纸过滤氯化银沉淀,用10%(v/v)盐酸冲洗表面皿、杯壁、沉淀和滤纸四次,滤液用100mL容量瓶接取,以10%(v/v)盐酸稀释至刻度,混匀,待测。[b]2. 结果与讨论2.1 仪器工作参数选择2.1.1 炬管位置的选择[/b] PEOptima 7300 DV全谱直读电感耦合等离子体原子发射光谱仪采用独特的双观测结构(轴向、侧向),其中轴向观测的检出限要比径向观测的低。由于样品中钒含量较低,所以本文选择炬管轴向观测方式进行测量。[b]2.1.2 分析谱线的选择[/b] 实验中对V的几条分析谱线,即290.880 nm,310.230 nm,309.310 nm,292.402 nm,311.071 nm和270.093 nm均进行了研究,实验中配制了浓度分别为0,2,5,10和20 ug×mL[sup]-1[/sup]的V标准工作溶液,分别在相同的实验条件下,在各个测定波长下绘制工作曲线。实验发现:几条谱线的线性都很好,灵敏度都能满足要求,故本文选择仪器推荐的测定波长290.880 nm。[b]2.1.3 发射功率的选择[/b] 用10 ug×mL[sup]-1[/sup]的V标准溶液,在波长290.880 nm处测定发射功率从900 W逐渐升至1400 W时谱线强度的变化。实验发现:随着发射功率的增加,谱线强度也增加。谱线强度较高,测定的灵敏度也较高,考虑到仪器硬件的自身物理性能,功率过高,易造成仪器性能的不稳定。故本实验方法选取的发射功率为1300 W。[b] 2.1.4 提升速度的选择[/b] 变化提升速度观察谱线强度的变化(以10ug.mL[sup]-1[/sup]的V标准溶液为测试溶液)。实验发现:随着样品提升速度的提高,谱线强度增加,在0.5~1.5mL×min[sup]-1[/sup]之间变化时增加显著,在1.5~2.0mL×min[sup]-1[/sup]之间的增幅趋缓,由于提升速度快消耗的样品量大,所以本文选取提升速度为1.5mL×min[sup]-1[/sup]。[b]2.2 背景干扰的扣除[/b] 本文利用仪器所带软件,在显示屏上显示V元素分析谱线的轮廓,用两点法选择最佳的背景扣除点,存入计算机中,当进行测定时,仪器会自动进行背景扣除。[b]2.3 共存离子的干扰[/b] 为了研究共存离子对钒测试准确度可能产生的影响,对一个银铜钒合金样品进行了定性和定量分析。发现样品中的主要存在元素是:Ag和Cu,还含有微量的K和Al。实验中根据样品中的元素相对含量,加入1000倍于钒含量的Ag和100倍于钒含量的Cu,测定其回收率为99.7 %~105.0 %,所以样品中的共存元素对钒的测试不造成干扰。[b]2.4 方法检出限和精密度[/b] 检出限(DL)定义为空白溶液测定标准偏差的3倍所对应的浓度。在优化过的实验条件下,对标准空白溶液测定11次后求得方法的检出限。用5 ug×mL[sup]-1[/sup]钒标准溶液连续测定11次计算相对标准偏差,求得精密度,其结果列于表2。[b] 表2 方法检出限和精密度(n=11)[/b] [table=569][tr][td] [align=center][b]样品溶液[/b][/align] [/td][td] [align=center][b]平均值[/b][/align] [/td][td] [align=center][b]标准偏差[/b][/align] [align=center][b] (mg/L)[/b][/align] [/td][td=2,1] [align=center][b]相对标准偏差(%)[/b][/align] [/td][td] [align=center][b]检出限(mg/L)[/b][/align] [/td][/tr][tr][td] [align=center]Blank[/align] [/td][td] [align=center]-0.161[/align] [/td][td] [align=center]0.0019[/align] [/td][td] [align=center]/[/align] [/td][td=2,1] [align=center] 0.0057[/align] [/td][/tr][tr][td] [align=center]5 ug/mL V[/align] [/td][td] [align=center]4.90[/align] [/td][td] [align=center]0.035[/align] [/td][td=2,1] [align=center]0.70[/align] [/td][td] [align=center]/[/align] [/td][/tr][/table][b]2.5 样品的检测[/b] 实验中采用两种不同的样品前处理方法(即:国家标准参考方法和建立的样品前处理新方法),分析了4份不同的银铜钒合金样品中的钒含量,测试结果见表3。[b] 表3 样品检测结果[/b] [table][tr][td] [align=center][b]样品编号[/b][/align] [/td][td] [align=center][b]依据国家标准方法的测试结果[/b][/align] [align=center][b](%)[/b][/align] [/td][td] [align=center][b]依据建立的新方法的测试结果[/b][/align] [align=center][b](%)[/b][/align] [/td][/tr][tr][td] [align=center]1[/align] [/td][td] [align=center]0.11[/align] [/td][td] [align=center]0.12[/align] [/td][/tr][tr][td] [align=center]2[/align] [/td][td] [align=center]0.12[/align] [/td][td] [align=center]0.12[/align] [/td][/tr][tr][td] [align=center]3[/align] [/td][td] [align=center]0.30[/align] [/td][td] [align=center]0.32[/align] [/td][/tr][tr][td] [align=center]4[/align] [/td][td] [align=center]0.12[/align] [/td][td] [align=center]0.13[/align] [/td][/tr][/table] 试验中对其中一个样品进行了加标回收试验,在样品中加入1 mL的钒标准溶液(r=100ug×mL[sup]-1[/sup]),然后按照相同的新建的样品前处理方法进行样品消解,所获得的试验结果见表4。[b] 表4样品加标回收试验结果[/b] [table][tr][td] [align=center][b]样品名称[/b][/align] [/td][td] [align=center][b]样品浓度[/b][/align] [align=center][b]ug×mL[sup]-1[/sup][/b][/align] [/td][td] [align=center][b]回收率[/b][/align] [align=center][b]%[/b][/align] [/td][/tr][tr][td] [align=center]样品[/align] [/td][td] [align=center]5.02[/align] [/td][td] [align=center]-[/align] [/td][/tr][tr][td] [align=center]加标样品[/align] [/td][td] [align=center]8.56[/align] [/td][td] [align=center]94.90[/align] [/td][/tr][/table][align=center] 从以上分析结果可以看出:所建立的新样品前处理分析方法与采用国家标准方法中推荐的样品前处理方法,进行银铜钒合金样品中钒含量的测试,所获得的实验结果没有明显区别。而本实验建立的新的样品前处理分析方法更加简便、快速和节约试剂成本,是银铜钒合金样品中钒含量测试的理想样品前处理分析方法。[/align][b]参考文献:[/b] 中华人民共和国国家标准GB/T15072.19-2008贵金属合金化学分析方法银合金中钒和镁量的测定电感耦合等离子体原子发射光谱法。2008年3月31日发布,2008年9月1日实施。中华人民共和国国家质量监督检验检疫总局、中国国家标准化管理委员会发布
我们公司是做废铜冶炼,化验室用原子吸收分析银时结果偏高,用铜矿石标准样(含银109克每吨)分析测得结果是132克左右,用铅锌矿石标准样(含银369克每吨),我们分析测得结果有时到440克了,我们溶解样品一般是称0.3~0.5克样品,先加15毫升左右盐酸溶解几分钟,,再加10毫升硝酸,0.3毫升溴溶解样品至近干,再加10毫升王水溶解至近干,加10毫升盐酸,加水至50毫升左右,煮沸,取下冷却,然后将溶液灌进100毫升容量瓶,至刻度摇匀,干过滤,进原子吸收,连同银标准溶液一起分析,四个标准0,0.5,1.0,2.0pg每毫升,线性系数一般在0.9994左右,分析结果和标准样比偏高,和外检一般也是280克的,也是偏高25克,请高手给以指点,问题在哪里。是标准溶液,还是样品处理。还有我们的样品中有些是铜粉,是别人处理线路板的,有很多塑料,所以我一般不用高氯酸处理样品,怕有危险的,我们样品来源很复杂的,有时会有各种杂质,看看处理样品除了高氯酸,还有更好的方法吗
摘 要:目的探讨饮用水中氯化氰的检验方法。方法采用吡啶一巴比妥酸光度法测定饮用水中氯化氰,对溶液的pH值、显色剂的配比、样品稳定性、氯胺T的加入量、显色反应时间和最大吸收波长等进行优化。结果该方法的线性范围为0~0.1μg/ml,相关系数为0.9994,回归方程为y=0.194x一0.033,检出限为0.003 μg/ml,测定下限为0.0l mg/L,相对标准偏差在1.5%-6.2%之间,回收率为92.9%一107.1%。结论该方法准确、灵敏、简便、快速,满足《生活饮用水卫生规范)(2001)的要求。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=102566]饮用水中氯化氰的吡啶一巴比妥酸分光光度测定法[/url]
在测铟消解时为什么加盐酸硝酸到近干,氯化铟不是易挥发吗,一直不太明白,这样不会导致结果偏低吗?急求大家的指点铟用王水溶样时在溶液中的状态到底是什么?谢谢
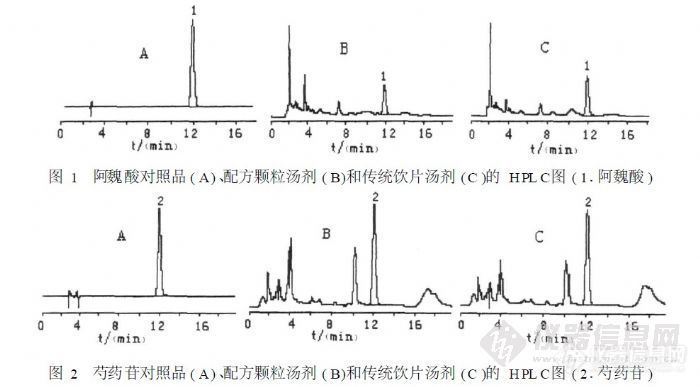
【作者】李媛 鲁定国 雷艳青 雷鹏 刘韶 李新中 【摘要】:目的:比较四物汤传统饮片汤剂与配方颗粒汤剂中阿魏酸、芍药苷的含量。方法:采用高效液相色谱法。色谱柱:Diamonsil C18(4.6 mm×250 mm,5μm);柱温:35℃;流速1 ml/min;测定阿魏酸流动相为甲醇-水-0.05%磷酸(40∶50∶10),检测波长为327 nm;测定芍药苷流动相为乙腈-水-0.05%磷酸(16∶74∶10),检测波长为230 nm。结果:自制配方颗粒汤剂中阿魏酸含量为8.91 mg/剂,芍药苷为83.57 mg/剂;传统饮片汤剂中阿魏酸含量为8.90 mg/剂,芍药苷为78.51 mg/剂;不同厂家配方颗粒汤剂中阿魏酸含量为3.36~7.67 mg/剂,芍药苷为48.13~67.52 mg/剂。结论:四物汤配方颗粒汤剂与传统饮片汤剂的色谱图基本一致,自制配方颗粒汤剂与传统饮片汤剂中阿魏酸和芍药苷含量基本相当,但不同厂家配方颗粒汤剂中存在显著性差异。【作者单位】: 中南大学湘雅医院 中南大学湘雅医院 中南大学药学院 中南大学湘雅医院 中南大学湘雅医院 中南大学湘雅医院 【关键词】: 高效液相色谱法 四物汤 传统饮片汤剂 配方颗粒汤剂 阿魏酸 芍药苷 http://ng1.17img.cn/bbsfiles/images/2012/07/201207310812_380724_2352694_3.jpg
跪求有没有人有“硫酸银、碳酸银”分析标准啊?
想请教一下各位老师,cod测定中。硫酸硫酸银溶液加硫酸银时加错了,一瓶硫酸500ml加了10克硫酸银,对cod的结果会有什么影响啊
大学时候有个同学,做硝酸银滴定时,不小心把硝酸银弄到手上了~(悲催)http://simg.instrument.com.cn/bbs/images/default/em09501.gif。听老师说了,硝酸银如果弄到衣服上,一段时间后会变黑。他就直接把手插到盐酸里了。。。http://simg.instrument.com.cn/bbs/images/default/em09501.gif,(当然盐酸浓度时比较稀的),之间他的手上立马出现了氯化银的白色沉淀......硝酸银没了,氯化银就洗不掉了......http://simg.instrument.com.cn/bbs/images/default/em09501.gif这就是他犯二的全过程.....后来,我问他手插到盐酸里的感觉如何?他说,手会微微痛.....然后.....就没有然后了~
COD中的硫酸硫酸银的配制,如果在一瓶浓硫酸中加入了10g硫酸银,会对COD产生什么影响?
我在用健合C18柱做维C银翘片的含量时,出现拖尾峰。流动相比例为甲醇:水:冰醋酸(20:80:0.5),又加在200ml流动相中加入0.5冰醋酸。结果没有改变。又用复方丹参的对照品较对下柱子,峰形正常。请问哪位行家能帮我个忙,我如何能得到对称峰?谢谢!!!
按《生活饮用水卫生规范》2001中铬酸钡分光光度法(热法)测水中硫酸盐。作出一组标准序列如下: 440nm波长 3cm比色皿 C(mg/L) 0 5 10 20 40 60 A 0.017 0.025 0.058 0.128 0.269 0.418 r=0.9976, 去掉0这点则有0.9999。显然0这点吸光值偏高,平常做过很多次,所有试剂重新配制,水用超纯水,情况都一样,也是标准空白偏高。采用420nm波长也一样。请各位高手指点一下到底是什么原因引起的?
我公司新购一台[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱仪[/color][/url],用优级纯硝酸或盐酸配置标液时,因酸中含一定量的金属元素,致使标液零点吸光度偏高,而样品本身待测元素含量偏低,测不出样品中待测元素含量。请问怎样减小标液零点吸光度?除更换酸外还有没有其他方法?
第九届原创离子色谱法测定饮料中的山梨酸、苯甲酸和糖精钠黄选忠 杜宏山 邹大喜(湖北兴山县疾病预防控制中心,443711)摘要 建立了用国产SH-AC-1型阴离子交换柱为分离柱,以1.5 mmol/LNa2CO3为淋洗液,流量为1.5mL/min,采用等度洗脱的方式测定食品中山梨酸、苯甲酸和糖精钠的方法,山梨酸、苯甲酸的线性范围为0~30.0mg/L,糖精钠的线性范围为0~15.0mg/L,方法应用于饮料等样品中山梨酸、苯甲酸和糖精钠的测定,其结果与气相色谱法相吻合,加标回收率分别为:97.9%~104.5%、96.3~100.9%和100.3%~104.5%,5次平行测定的相对标准偏差分别为:1.68%~3.92%、2.32~4.08%和2.02%~3.95%(n=5),按样品稀释50倍计方法的检出限分别3.5、2.5和1.5 mg/L。关键词 离子色谱法,饮料,山梨酸,苯甲酸,糖精钠食品中山梨酸、苯甲酸和糖精钠的测定方法国家标准推荐的主要有气相色谱法和高效液相色谱法等,其中,气相色谱法样品前处理方法繁杂,而高效液相色谱法虽然样品前处理方法简单但仪器价格偏贵基层实验室少有配置,使其应用受到限制。离子色谱法以其灵敏、快速、试剂消耗少、无环境污染等优点而备受广大分析工作者的青睐,已应用于食品中山梨酸、苯甲酸和糖精钠等多种添加剂的分析,但这些方法大多使用的是进口色谱柱并采用KOH淋洗液梯度洗脱方式进行测定,而进口色谱柱特别是具有梯度洗脱功能的离子色谱仪的价格普遍偏贵,目前基层实验室装备较少难以应用,因此研究用国产普通色谱柱和普通色谱仪测定食品中山梨酸、苯甲酸和糖精钠是一项有意义的工作,本工作研究了用国产普通色谱柱和色谱仪测定食品中山梨酸、苯甲酸和糖精钠的各种条件和可行性,结果表明,以1.5 mmol/LNa2CO3溶液作淋洗液,流量为1.5ml/min,用SH-AC-1型阴离子交换柱为分离柱,采用等度洗脱的方式可使山梨酸、苯甲酸和糖精钠较好分离,各组分的峰面积与其浓度在一定的范围内具有良好的线性关系,且各组分的保留时间在14 min以内,具有分析应用价值。据此,建立了用国产SH-AC-1型阴离子交换柱为分离柱,以1.5 mmol/LNa2CO3为淋洗液等度洗脱的方式测定食品中山梨酸、苯甲酸和糖精钠的方法,山梨酸、苯甲酸的线性范围为0~30.0mg/L,糖精钠的线性范围为0~15.0mg/L,方法应用于饮料等样品中山梨酸、苯甲酸和糖精钠的测定,获得了满意的结果。1、实验部分1.1主要仪器CIC-200型离子色谱仪(青岛盛翰色谱公司),抑制器:检测器:电导检测器,定量环体积为25μL;分离柱,SH-AC-1型阴离子交换柱(250×4.0mm i.d,青岛盛翰色谱公司,批号:1404016);1.2 仪器工作条件 柱箱温度35℃,电流:50mA。1.3主要试剂山梨酸、苯甲酸和糖精钠标准溶液:1000 mg/L,按照文献方法配制。临用时用纯水将各种标准溶液稀释成含山梨酸、苯甲酸各250.0 mg/L、 糖精钠125.0mg/L的混合标准应用液;75 mmol/LNa2CO3淋洗液贮备液,临用时用纯水稀释50倍使用。实验所用试剂均为AR及以上级,实验用水为超纯水(18.25ΜΩ·cm)。1.4 实验方法1.4.1标准曲线的绘制 取0.10、0.20、0.60、1.00、2.00和3.00 mL混合标准液于25mL容量瓶中加纯水至刻度,混匀,配制成含山梨酸、苯甲酸1.0、2.0、6.0、10.0、20.0、 30.0mg/L和糖精钠0.5、1.0、3.0、5.0、10.0和15.0mg/L标准系列,各进样1 mL上机(测试条件为,柱箱温度:35℃,电流:50mA;淋洗液:1.5mmol/L Na2CO3溶液;淋洗液流量:1.5mL/min;量程:1档)测定各成份峰面积(S),以S对浓度绘制工作曲线。1.4.2样品测定 将样品稀释50倍经0.45μm滤头过滤后进样1 mL上机(条件同1.4.1)测定各成份峰面积(S),以标准曲线法定量。2、结果与讨论2.1 Na2CO3浓度的选择 在高效液相色谱法中选择适当的淋洗液是改善分离度(R)的有效方法,为保证山梨酸、苯甲酸和糖精钠的有效分离,同时当以Na2CO3作淋洗液时,NO3-的保留时间与苯甲酸的保留时间比较接近,考虑到样品中可能存在的NO3-对苯甲酸测定的影响,为此进行了Na2CO3淋洗液浓度的选择实验,结果见表1。从表1可见,当Na2CO3浓度在1.0~表1 Na2CO3浓度对分离度和保留时间的影响(流量1.0 ml/min) 组分/浓度(mg/L) 1.0mmol/L 1.5mmol/L 2.0mmol/L 保留时间(min) R 保留时间(min) R 保留时间(min) R 山梨酸(20) 11.487 2.68 9.630 2.66 8.438 2.29 苯甲酸(20) 19.09 1.46 15.598 1.16 13.463 1.03 NO3-(10) 22.509 1.78 17.870 1.63 15.212 1.60 糖精钠(10) 26.048 / 20.645 / 17.543 / [/td
离子色谱法测定饮料中的山梨酸、苯甲酸和糖精钠黄选忠 杜宏山 邹大喜(湖北兴山县疾病预防控制中心,443711)摘要 建立了以青岛盛翰色谱公司生产的SH-AC-1型阴离子交换柱为分离柱,以1.5 mmol/LNa2CO3为淋洗液,流量为1.5mL/min,采用等度洗脱的方式测定食品中山梨酸、苯甲酸和糖精钠的方法,山梨酸、苯甲酸的线性范围为0~30.0mg/L, 糖精钠的线性范围为0~15.0mg/L,方法应用于饮料等样品中山梨酸、苯甲酸和糖精钠的测定,其结果与气相色谱法相吻合,加标回收率分别为:97.9%~98.4%、96.5~96.8%和100.6%~104.4%,5次平行测定的相对标准偏差分别为:1.68%~3.92%、2.32~4.08%和2.02%~3.95%(n=5),按样品稀释50倍计方法的检出限分别3.5、2.5和1.5 mg/L。关键词 离子色谱法,饮料,山梨酸,苯甲酸,糖精钠食品中山梨酸、苯甲酸和糖精钠的测定方法国家标准推荐的主要有气相色谱法和高效液相色谱法等,其中,气相色谱法样品前处理方法繁杂,而高效液相色谱法虽然样品前处理方法简单但仪器价格偏贵基层实验室少有配置,使其应用受到限制。离子色谱法以其灵敏、快速、试剂消耗少、无环境污染等优点而备受广大分析工作者的青睐,已应用于食品中山梨酸、苯甲酸和糖精钠等多种添加剂的分析,但这些方法大多使用的是进口色谱柱并采用KOH淋洗液梯度洗脱方式进行测定,而进口色谱柱特别是具有梯度洗脱功能的离子色谱仪的价格普遍偏贵,目前基层实验室装备较少难以应用,因此研究用国产普通色谱柱和普通色谱仪测定食品中山梨酸、苯甲酸和糖精钠是一项有意义的工作,本工作研究了用国产普通色谱柱和色谱仪测定食品中山梨酸、苯甲酸和糖精钠的各种条件和可行性,结果表明,以1.5 mmol/LNa2CO3溶液作淋洗液,流量为1.5ml/min,用SH-AC-1型阴离子交换柱为分离柱,采用等度洗脱的方式可使山梨酸、苯甲酸和糖精钠较好分离,各组分的峰面积与其浓度在一定的范围内具有良好的线性关系,且各组分的保留时间在14 min以内,具有分析应用价值。据此,建立了以青岛盛翰色谱公司生产的SH-AC-1型阴离子交换柱为分离柱,以1.5 mmol/LNa2CO3为淋洗液等度洗脱的方式测定食品中山梨酸、苯甲酸和糖精钠的方法,山梨酸、苯甲酸的线性范围为0~30.0mg/L, 糖精钠的线性范围为0~15.0mg/L,方法应用于饮料等样品中山梨酸、苯甲酸和糖精钠的测定,获得了满意的结果。1、实验部分1.1主要仪器CIC-200型离子色谱仪(青岛盛翰色谱公司),抑制器:检测器:电导检测器,定量环体积为25μL;分离柱,SH-AC-1型阴离子交换柱(250×4.0mm i.d,青岛盛翰色谱公司,批号:1404016); 1.2 仪器工作条件 柱箱温度35℃,电流:50mA。1.3主要试剂山梨酸、苯甲酸和糖精钠标准溶液:1000 mg/L,按照文献方法配制。临用时用纯水将各种标准溶液稀释成含山梨酸、苯甲酸各250.0 mg/L、 糖精钠125.0mg/L的混合标准应用液; 75 mmol/LNa2CO3淋洗液贮备液,临用时用纯水稀释50倍使用。实验所用试剂均为AR及以上级,实验用水为超纯水(18.2ΜΩ·cm)。1.4 实验方法1.4.1 标准曲线的绘制 取0.10、0.20、0.60、1.00、2.00和3.00 mL混合标准液于25 mL容量瓶中加纯水至刻度,混匀,配制成含山梨酸、苯甲酸1.0、2.0、6.0、10.0、20.0、 30.0mg/L和糖精钠0.5、1.0、3.0、5.0、10.0和15.0mg/L标准系列,各进样1 mL上机(淋洗液:1.5mmol/L Na2CO3溶液;流量:1.5mL/min;量程:1档)测定各成份峰面积(S),以S对浓度绘制工作曲线。1.4.2 样品测定 将样品稀释50倍经0.45μm滤头过滤后进样1 mL上机(条件同1.4.1)测定各成份峰面积(S),以标准曲线法定量。2、结果与讨论2.1 Na2CO3浓度的选择 在高效液相色谱法中选择适当的淋洗液是改善分离度(R)的有效方法,为保证山梨酸、苯甲酸和糖精钠的有效分离,同时当以Na2CO3作淋洗液时,NO3-的保留时间与苯甲酸的保留时间比较接近,考虑到样品中可能存在的NO3-对苯甲酸测定的影响,为此进行了Na2CO3淋洗液浓度的选择实验,结果见表1。从表1可见,当Na2CO3浓度在1.0~表1 Na2CO3浓度对分离度和保留时间的影响(流量1.0 ml/min)组分/浓度(mg/L)1.0mmol/L1.5mmol/L2.0mmol/L保留时间(min)R保留时间(min)R保留时间(min)R山梨酸(20)11.4872.689.6302.668.4382.29苯甲酸(20)19.091.4615.5981.1613.4631.03NO3-(10)22.5091.7817.8701.6315.2121.60糖精钠(10)26.048/20.645/
作者:金智利; 袁文婧;哈药集团三精制药股份有限公司;摘要:目的:建立高效液相色谱法测定金银花连翘提取物中绿原酸、咖啡酸的的含量。方法:采用迪马(钻石)C18(250mm×4.6mm,5μm)色谱柱,流动相为甲醇-水-冰乙酸(20:80:1),流速1.0ml/min,检测波长:324nm。结果:金银花连翘提取物中绿原酸、咖啡酸能达到有效分离,绿原酸在9.44μg/ml~141.60μg/ml范围内,浓度同峰面积呈良好的线形关系,仪器精密度RSD为0.03%,方法的重复性RSD=0.24%。咖啡酸在5.36μg/ml~80.40μg/ml范围内,咖啡酸峰面积值与浓度有良好的线性关系,仪器精密度RSD为0.03%,方法的重复性RSD=0.33%。结论:该方法简便、准确、可行。可用于金银花连翘提取物的质量控制。
乳酸(菌)饮料是饮料,酸奶是天然食品,乳酸(菌)饮料与酸奶是完全不同的两种食物。凡是有“饮料”二字的食品,医生或是营养专家都不会建议长期饮用。 具体来说,酸奶是由优质的牛奶经过乳酸菌发酵而成的,本质上属于牛奶的范畴,保存了鲜奶中所有的营养素,含有丰富的蛋白质、脂肪、矿物质。 而乳酸(菌)饮料,大部分是以鲜奶或奶粉为原料,经乳酸菌培养发酵制得,再在制得的乳液中加入糖、各种香料以及其他添加物质而最终制成。在稀释了酸奶的同时,根据各厂家的产品不同,还添加了复杂多样的成分。 区分是酸奶还是饮料,主要要看蛋白质含量。 根据国家标准,酸奶和含乳饮料的包装上都应标明产品成分和配料。酸奶的配料表中,蛋白质含量标示不应低于2.3%,牛奶3.0%,乳酸(菌)饮料蛋白质含量一般只有1%。 酸奶以及乳酸(菌)饮料对人体肠道的健康作用,主要区别于真正作用于人体肠道内的益生菌数量。 就其本身含有的益生菌数量来说,从高到低含量排序为酸奶、乳酸菌饮料、乳酸饮料。乳酸菌和乳酸饮料的区别就在于是否含有益生菌。 有些益生菌饮料广告,宣传语建议,每天饭后来一瓶,可帮助消化促进肠道蠕动。对此,专家指出,不建议依靠益生菌饮料来补充人体肠道益生菌数量,因为它的效果微乎其微。 益生菌饮料,根据各产品制作工艺和保存方式的不同,本身含有一定量的活性益生菌,在运输、保存过程中,会有一部分益生菌死亡,失去活性。 喝到人体内,进入胃部,由于胃酸的刺激,又有绝大部分活性益生菌死亡,变成死菌。 但是,也不排除有能“快速通过”胃部,逃脱胃酸的杀害到达人体肠道的少量益生菌确实能发挥对人体的健康作用。
我司是生产成品锡膏,有合金锡银铜,锡铋银,锡铋类锡膏,在近几个月前处理样品总会出现沉淀. 按照下列方法处理样品还会出现沉淀物,希望大家一起来讨论 对于纯水后20%酸度指的是什么意思 是浓度比还是体积比, [font=金桥繁宋体]• [/font][font=金桥繁宋体]锡和锡合金:金属锡溶于浓盐酸、热的浓硫酸和王水。锡易生成白色不[/font][font=金桥繁宋体]溶性偏锡酸沉淀,锡合金用盐酸+硝酸(9+1)溶解,加入超纯水稀释至[/font][font=金桥繁宋体]20%酸度溶液 [/font][font=Wingdings]l[/font][color=black][font=华文行楷]有色金属前处理方法[/font][/color]
近来在做枯矾中硫酸铝钾的含量测定,用药典2010年版的方法。发现如果直接灰化什么都不加,得到的含量值偏大,而如果前处理加入硫酸炭化后再灰化,值会偏小。不知道该不该加硫酸,加硫酸是参照炽灼残渣项下操作规范做的。而且,不应该是值越大灰化的才越完全吗,希望能帮忙解答下,谢谢!

测COD用的风冷装置,同一个水样,对比别的实验室水冷出来的结果偏低,风冷测出来578,别的实验室水冷测出来643,另一个水样风冷测出来278,另一个实验室水冷做出来301,我们是把硫酸硫酸银直接加入到加热管,这个会不会是误差存在的主要原因。但是我之前用水冷和风冷做同一个水样,出来的结果是151和149,差别并不大,请教各位大神帮我分析一下。我们用的下图风冷装置。[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2018/08/201808291634156003_8453_3466237_3.png[/img]
矿石中铅锌金银含量测定遇到的问题测定矿石中铅锌金银含量的时候结果都比别的实验室测出的偏低,实在找不出原因,专家们给点建议哈。测金用的活性炭吸附法;测银用盐酸硝酸高氯酸分解,在1+9盐酸介质中测定;铅锌用王水溶解,这都是从冶金分析手册中找的方法,不知道为什么和别的实验室测出的结果相差较大
我配制了硫酸-硫酸银溶液,用来作COD检测的,是用浓硫酸溶解硫酸银,没有用水,但放置一段时间后就不能用了,这是怎么回事啊?[em06] [em28]
各位老师好,水质银曲线是质控样都是新配的,曲线做出来 0.9999,但是我的低中高浓度质控样做出来都不在质控范围内,而是偏低,这是怎么回事呢,还有就是所有的质控我做出来都偏低,从来没有偏高过,不知道为什么?我也排查了一些问题,但都没用,做出来还是低,希望大家帮我分析一下,看看哪里出问题了

作者:李昂; 胡翮; 郭丙炎;(湖南省湘潭市药品检验所;)摘要:目的用反相高效液相色谱(RP-HPLC)法测定盐酸丁卡因的含量。方法色谱柱为Diamonsil C18柱(150mm×4.6mm,5μm),流动相为水-甲醇-乙腈(45∶45∶10,v/v,含0.02%庚烷磺酸钠,0.34%磷酸二氢钾,三乙胺调pH至7.0),流速为1.0mL/min,检测波长为314nm,柱温为室温。结果盐酸丁卡因质量浓度在5.034~161.1μg/mL范围内与峰面积线性关系良好,回归方程为Y=5.6572×105X-9.2039×105(r=0.9999),平均回收率为100.76%,RSD为0.41%(n=6)。结论RP-HPLC法简便、可靠、准确,可作为盐酸丁卡因注射液的含量测定方法。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208271103_386360_1606903_3.jpg
做银矿石银含量的时候,我的师傅发现有机物含量较高,他先加的硝酸,然后加的盐酸,可是如果矿样含硫高的话,浓硝酸和硫反应成了硫酸,那么是不是就间接的生成了硫酸银和硫化银了,那在最后加硫脲的时候还能反应成硫脲银了么?
有谁做银耳的米酵菌酸?条件是什么啊?
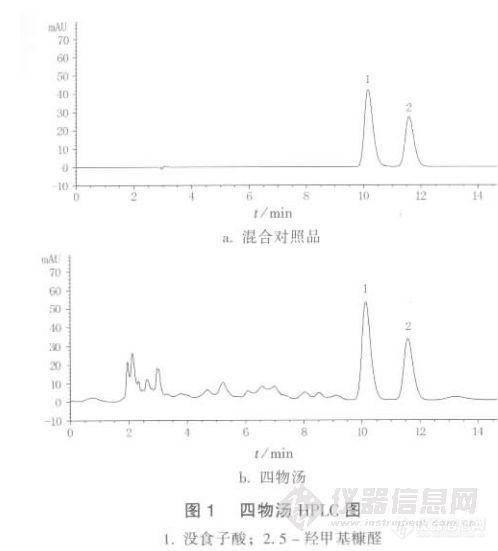
【作者】:雷 鹏 ,李 媛,李新中,黄 义【摘要】:目的 建立 Q7W- 法同时测定四物汤中没食子酸、D E 羟甲基糠醛含量的方法,并比较四物汤传统饮片汤剂与配方颗粒汤剂中没食子酸、D E 羟甲基糠醛的含量。方法 1(&8*+0(, -G# 色谱柱 H !B " 88 X ;D 88,D !8J ;流动相:甲醇 E
硫酸-硫酸银溶液撒在通风橱桌面上能用氯化钙处理吗?如果不能该怎么办?以后配制硫酸-硫酸银溶液时该注意什么?这次是用烧杯配硫酸-硫酸银溶液,在搅拌溶解时烧杯底漏了,量是1L的,所以想问问怎么解决







 我要推广仪器
我要推广仪器
 下载APP
下载APP