请问1,5-二巯基-1,3,4-噻二唑、2-取代烷基-5-巯基-1,3,4-噁唑啉的核磁谱图如何解析?

http://ng1.17img.cn/bbsfiles/images/2012/09/201209270900_393235_2351433_3.jpg聚芳噁二唑(Poly-1,3,4-oxadiazole,简称POD),各位高手能否帮小妹分析一下,谢谢了!!
最近看到仪器信息网上的一个帖子,关于安捷伦二噁英论坛上的 关于持久性有机物的研讨会中提到qqq做二噁英类,想问一下各位老师有没有知道国内外的方法中用qqq做二噁英的相关方法呢。
[em09504]测定磷酸三丁酯,以邻苯二甲酸二丁酯做内标物,以二甲苯作溶剂,峰会重叠吗?http://www.instrument.com.cn/bbs/shtml/20030612/218050/以二甲苯作溶剂,大家觉得按上面的帖子可行吗?邻苯二甲酸二丁酯使用分析纯的可以吗?还是要用色谱纯的?
求助,哪家可以做二噁英的仪器计量啊,赛默飞的磁质谱
求助 邻苯二甲酸二乙酯在液谱上做内标物的应用实例谢谢!!!
谁做二噁英检测? 有EPA相关标准分享一下?如EPA 1613 等。
做水质苯系物可以直接用二硫化碳做标曲吗?买的二硫化碳标液说明书上说可以用于做水质苯系物
二硫化碳中苯系物用什么做内标,苯系物是质控二硫化碳中7种苯系物 苯,甲苯 ,对间邻二甲苯,乙苯,苯乙烯我用的气相色谱仪 进样梁很不准确 所以需要用内标,但不知道选什么当内标,大家帮下忙,谢谢!!
做苯系物时突然想到的,标准上要求使用的毛细管柱是PEG-20M,强极性柱。我记得物质在极性柱上的出峰顺序是按照极性由弱到强来的,我记得苯的极性是甲苯二甲苯的吧。那按照正常的出峰顺序应该是先出二甲苯,然后是甲苯,然后是苯。为什么标准上给出的顺序是苯,甲苯,二甲苯。我实际做确认时也确实是苯先出,是二硫化碳对苯系物的极性造成某些影响导致的吗?
Agilent 6890/5973N顶空进样,做二噁烷残留,检测限最低只能做到2.5个PPM,再低一点就不行了,可是看网上有的人可以作到0.2-0.3个PPM,不知道是哪方面的原因,知道的达人告诉一声啊,多谢了!
买的二硫化碳中八种苯系物说明书是说适合11890-89,这样直接做曲线可以吗?或者把配好的标液再加到分液漏斗里模拟一下萃取流失?还是直接用甲醇做标曲。
最近有盲样考核,之前做的都是的二硫化碳中苯系物,没有甲醇中曲线
本人对不确定度还不太懂,现在要做二噁烷的不确定度,用的方法是国食药监许456号附件7《化妆品中二噁烷的检测方法》,不知道如何入手,不知哪位大侠做过这类的模板
想请教大家,用二硫化碳解析做苯系物的色谱瓶能重复利用吗?因为我们用量还挺大,而这种样品瓶还挺贵,但是二硫化碳的毒性又很大,领导的意思要重复利用,但是对身体危害大啊,要是能清洗怎么清洗呢?
请教诸位同仁:多谢赐教 有没有做食品中二噁英的同仁,GB5009.205-2007要求使用高分辨MS,分辨率10000,那么普通的四级杆GCMS或者GCMSMS是否不能满足要求? 什么形制的GCMS可以满足要求?磁质谱么?或者TOF?有没有商品仪器?
各位大神,想问一下,用DB-FFAP的柱子做二硫化碳苯系物设置什么仪器参数比较好。我的进样口150,检测器220,柱箱是程序升温,60度保持1min以5℃/min的速率升到95度,分流比10:1做出来的峰特别差,还分不开,大家都用的什么条件做的
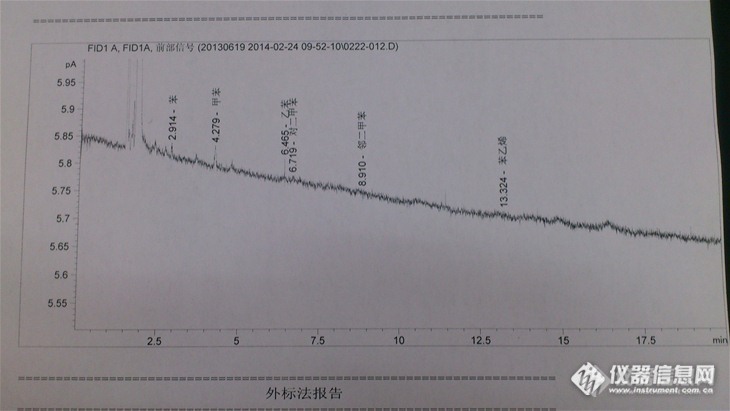
活性炭吸附二硫化碳解析做空气中的苯系物,仪器为GC-FID,DNP填充柱和PEG-20M毛细管柱,由于仪器好久没用,如图,做出来的基线老往下掉,噪声很大,这是什么原因?是不是需要老化柱子了,还有什么问题呢? http://ng1.17img.cn/bbsfiles/images/2014/02/201402241224_490953_2785618_3.jpg
做标准曲线是甲醇中的半挥发性有机物,而我做水样是用二氯甲烷来萃取半挥发性有机物的,请问这样做会不会影响水样中半挥发性有机物的含量?这个问题想了很久都想不通,请各位大侠帮忙确认可以吗?
那位大虾能赐教一下,我们想采集江水做其中的二噁英,谁那儿有采集的相关方法呀?求助求助!
做苯系物的曲线少出了一个邻二甲苯的峰,这是为什么?用的仪器是GC9790[img]https://ng1.17img.cn/bbsfiles/images/2018/12/201812251411101237_9662_3485882_3.png[/img]
我们是拿二硫化碳的苯系物标准和考核样直接进样做的,最近上面说这个不规范,请问大家都是用什么方法做的呢?
[color=#444444]几个月前买了二溴新戊二醇做实验,感觉原料不对劲,询问厂家,他们说没有问题。[/color][color=#444444]于是打了质谱,我们学校的质谱是电喷雾类型的,二溴新戊二醇的分子量是261.94,在质谱上可以找到262或者263,和303的峰,但比较矮(最近质谱仪可能出了问题,所以也可能是要多加1或2)。打了红外,与网上找到的红外谱图对应的很好。但是,质谱谱图上没有Br的同位素峰。大家都知道,有机物含有两个溴时,是1比2比1,含有一个溴时是一比一,而我的质谱谱图上只有一个峰。头痛啊。[/color][color=#444444]哪位经验丰富的大虾懂得其中的缘由啊。难道真的是原来出了问题?[/color]
做药物的有机溶剂残留,用二甲基亚砜做溶解药品的溶剂,二甲基亚砜本身有好多小杂质峰干扰药物中的待测有机溶剂的测定,有谁用过二甲基亚砜做过溶解样品的溶剂的,在2-5min是不是也有好多小杂质峰干扰测定?换了好几个厂家的二甲基亚砜了,或多或少都有干扰,如何处理?还有什么其它溶剂可用没有,甲醇、甲苯、正丁醇、正己烷做为溶剂药物都不溶解(我测药物中乙醇,乙酸乙酯、二氯甲烷、四氢呋喃、NN-二甲基甲酰胺的残留)
请问分析甲醇中的苯系物要用什么做固定相的色谱填充柱?我们公司先买了一根适合分析二硫化碳中的苯系物,一作甲醇中的苯系物就只有溶剂峰。
[size=5][font=FangSong_GB2312]目前想向广大的全部的所有的实验室监测站的朋友咨询下关于二噁英的对这类产品有哪些的设备?有没有具体的资料或者设备给我看看,我想了解类似的设备,不管进口的国产的二噁英设备,分析的采样的等都可以,还有目前那个实验室有类似的设备或者做二噁英的分析,谢谢大家了。我也是紧急求用。希望大家多多帮忙。[/font][/size]
做HJ605土壤时样品中的二溴氟甲烷替代物偏大很多,空白也是(加入五十测到了一百多),而其他两个替代物是正常的,但是在加标样品种三个替代物都是正常的!用的同一个使用液,求助大神!
气相测3.5-二甲氧基苯甲酸甲酯要用什么标准物做外标
我前一段时间做了一些含二茂铁化合物及含铬的配合物的核磁实验,图谱很清晰,只是二茂铁化学位移值及含铬的配合物的苯环化学位移值往低场移动。请问各位,是否可以作这样的实验?做这些实验对机器有何不利影响?
做邻苯二甲酸盐有机物含量测试大家都习惯用那些试剂做萃取?具体有哪些要求?杂质少的有哪些?







 我要推广仪器
我要推广仪器
 下载APP
下载APP