鸡蛋中四环素三氯乙酸溶液怎么配

刚入手axios max,看各位讨论粉末塑料环法做样重复性perfect,特请教这个塑料环长的什么样呢?请手上有货的亲们上上图呀。这个塑料环应该不是一次性使用的吧?随机买的这种应该不能压片的呀。[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/03/201903271837470953_5975_1731308_3.png[/img]

试环-试块滑动磨损试验方法是材料类评定摩擦磨损性能的试验方法,金属材料参考国标《GB/T 12444-2006 金属材料磨损试验方法 试环-试块滑动磨损试验》,塑料及塑料基复合材料参考国标《GB/T 3960-89 塑料滑动摩擦磨损试验方法》。 国标GB/T 12444-2006 试验结果处理时指出:“在块试样磨痕中部及两端(距试样边缘1mm处)测量磨痕宽度,取3次测量平均值作为一个试验数据,标准尺寸试样三个位置的磨痕宽度之差大于平均宽度值20%时,试验数据无效”。国标GB/T 3960-89也同样明确指出:“本标准以磨痕宽度来表征磨损量。测量三点,取平均值,各点之差不得大于1mm。”换句话说,就是试环-试块滑动磨损试验方法试验结束,试块的整体磨痕宽度须在标准规定的范围之内,否则试验无效。可见,试验结束后磨痕的状态直接表征试验的有效性。 同时,若试验结束后的磨痕状态不规则,也同样会在一定程度上影响磨损量的结果,摩擦系数也必然会受到一定程度的影响。虽然,标准GB/T 12444-2006有说明:“由于试块在磨损中受材料转移、氧化膜行程、润滑剂渗透等影响,试块的磨损量一般不用质量损失计算。”但是,对部分材料来说,在一定条件下做磨损对比性试验,还是有一定的参考意义。那么磨痕的不规则性是怎样造成的,又与哪些方面的因素有关系? 如下图,为在济南益华摩擦学测试技术有限公司生产的设备MRH-3型 高速环块摩擦磨损试验机上作的一组比照试验。照片为试验结束后的磨痕状态。观察照片可知,图2接近于标准磨痕状态,整体宽度、状态相对比较均匀、规则,而图1磨痕形状为梯形偏离标准要求的磨痕状态,更有严重偏离标准要求的结果接近于三角形。http://ng1.17img.cn/bbsfiles/images/2016/07/201607050952_599222_3080793_3.jpg http://ng1.17img.cn/bbsfiles/images/2016/07/201607050952_599224_3080793_3.jpg 图1 图2 在这里济南益华摩擦学测试技术研究所经多年客户委托试验经验作简要分析。 第一,国标GB/T 12444-2006、GB/T3960-89中,都明确规定了试验用试样尺寸及精度。一旦试样尺寸加工不合格,如摩擦表面处理不一致、不平整、试块表面不平行、试环内孔及表面精度达不到要求等都易导致如图1的试验结果,更甚是角度更大的梯形磨痕,严重影响试验结果评价。 第二,卡具加工精度。单单试样的精度达到要求,若块试样卡具槽不平行或主轴精度等达不到要求,与试块、试环配合不好,也同样会直接影响试验结果磨痕的规范性。 第三,设备精度。除了试样、卡具的加工精度,磨痕的规范性与设备的精度也是密不可分的。设备精度不达标,如试验相关机械部件整体装配精度、本身的精度(比如弹簧加载系统的精度、试样装卡系统精度等)皆直接影响着磨痕的规范性。 第四,人员操作因素。比如设备本身加载系统中,加载过程中试样块部分是可以自动校正试块与试环接触位置角度的,若操作人员采用的试验力值较大时,试验前直接将试验力加载至设定值,然后再启动试验,这样不仅容易造成磨痕的不规范性,更容易导致试验无法正常进行,可能在试验进行一定时间后由于摩擦力大或振动大致试验停止。 除此之外,磨痕的规范与否与材料本身也有直接的关系。如果试样块或试环接触面内部组织分布不均匀,造成摩擦接触位置相对一边硬一边软或是一边自润滑效果好一边自润滑效果不好等类似现象,也极易导致磨痕的不规范。 针对国标GB/T 12444-2006、GB/T 3960-89来说,磨痕的不规范直接导致试验的无效。只有正确认识到影响磨痕的试验因素,正确改进并使设备精度、试样加工精度皆达到标准及行业要求,提高自身测试技术水平,才能更好的提高试验的有效性,得出更有意义的研究结论,对材料作出更可靠的评价。
四环素制药废水一般怎么模拟?
本人是质谱新手。仪器 AB 5500。最近在优化四环素类质谱参数,在第一步找母离子的时候目标质量数的响应值只有3e5,而且还不是响应值最高的,有其他的质量数跟目标质量数的响应值一样。按照工程师的培训的要求母离子响应值应该在1-3e6。 最近一次调谐是在两个星期前。各位老师帮忙分析一下是哪的问题?浓度是100ng/ML,溶剂是1:1甲醇水 标注品是固体 盐酸金霉素、盐酸土霉素、盐酸强力霉素、盐酸四环素。
附件为ISO14001环境管理基础知识培训材料,供大家参考,学习。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=79156]ISO14001环境管理基础知识培训[/url]目 录1. ISO14001基础知识1.1 ISO14000系列标准中的主要标准1.2 ISO14001的产生1.3 ISO14001关注的焦点1.4 ISO14001的管理核心1.5 ISO14001的基础1.6 两个重要的名词1.7 企业中可能存在的七种类型的环境因素1.8 企业建立ISO14001环境管理体系可产生的效应1.9 ISO14001:2004标准的主要内容及其相关间关系2. 本公司环境管理体系对全体员工的基本要求2.1 废弃物管理2.2 能源资源管理2.3 应急准备与响应2.4 消防安全知识2.5 废水废气噪音治理知识3. 公司环境方针目标指标介绍3.1 环境方针3.2 环境目标指标4. 环境因素培训
0.05mol/L(50mmol/L)的磷酸盐缓冲溶液(pH=8)怎么配?有具体的配制方法更好,也可以说一下配制思路。PS:我看到磷酸盐缓冲溶液一般是用磷酸二氢钾和磷酸氢二钠配,为什么不干脆用两个钠盐呢(即磷酸二氢钠和磷酸氢二钠)?如蒙赐教,万分感谢!
如题,请问哪里可以购买压片机模具用的塑料环,现在用的硼酸感觉比较麻烦,想用塑料环试试效果
我要配的是200毫升0.4mol/L硼砂-硼酸缓冲溶液,PH=9.3,请问该怎么配?
各位大侠,请问有没有关于环氧模塑料水解氯的测试方法或者国标的?或者是电工用双酚A环氧树脂中可水解氯的测定,急用,多谢喽
我现在要配制盐酸土霉素,盐酸金霉素,盐酸四环素,盐酸多西环素,这四种物质除了盐酸金霉素是微溶于水的,其他三种都是溶解于水,可是我配制1000mg/L,发现还是有没有溶解的,在配制的过程中感觉盐酸多西环素还是比较容易溶解于水的,但那三种配的过程中就有悬浮的放置一个晚上后好多沉淀有没有哪位大侠做过这方面研究啊,指点一下,十分感谢,十分感谢我用的液相色谱测峰,之前配过盐酸四环素,盐酸金霉素,土霉素,多西环素,也是溶解的不好,所以才改全是盐酸盐,但溶解的还是不好做标线用甲醇配的,溶解的还可以,出峰也不错,但在水里我是怎么也溶解不了
[B]动物源性食品中四环素、沙星类[/B] 残留量的快速测定方法1 范围本方法规定了动物源性食品中四环素类、沙星类高效液相色谱的快速测定方法。本方法适用于动物源性食品中四环素类、沙星类高效液相色谱的快速测定。2.1 原理试样中的残留物经四环素类、沙星类快速检测前处理试剂盒处理,样液经四环素类专用层析柱净化、浓缩用高效液相色谱检测,外标法定量。2.2 试剂和材料除另有规定外,所有试剂均为分析纯,水为重蒸馏水。2.2.1 乙腈:色谱纯。2.2.2 甲醇:色谱纯。2.2.3 三乙胺(分析纯)2.2.4 磷酸(85%)(分析纯)2.2.5 磷酸氢二钠:优级纯。2.2.6 乙二胺四乙酸二钠。2.2.7 柠檬酸:分析纯。2.2.8 磷酸氢二钠溶液:0.2mol/L。称取28.41g磷酸氢二钠,用水溶解,定容至1000mL。2.2.9 柠檬酸溶液:0.1mol/L。称取21.01g柠檬酸,用水溶解,定溶至1000mL。2.2.10 Mcllvaine缓冲溶液:将1000mL0.1mol/L柠檬酸溶液与625mL0.2mol/L磷酸氢二钠溶液混合。2.2.11 Na2EDTA-Mcllvaine缓冲溶液:0.1mol/L。称取60.5g乙二胺四乙酸二钠放入1625mLMclllvaine缓冲溶液中,使其溶解,摇匀。2.2.12 标准品: 土霉素、四环素、金霉素、强力霉素、氧氟沙星、诺氟沙星、环丙沙星、单诺沙星、恩诺沙星纯度大于98 %。2.2.13 标准贮备溶液:分别称取土霉素、四环素、金霉素、强力霉素、氧氟沙星、诺氟沙星、环丙沙星、单诺沙星、恩诺沙星各10mg,用甲醇溶解并定溶于100 mL棕色容量瓶中,配制成100 µ g/mL的标准贮备液,置于-20℃保存,有效期三个月。2.2.14 混合标准工作溶液:用流动相稀释标准贮备溶液,配制成土霉素、四环素、金霉素、强力霉素、氧氟沙星、诺氟沙星、环丙沙星、单诺沙星、恩诺沙星均为10 µ g/mL的混合标准溶液。0~4℃避光保存。2.2.15 四环素、沙星类快速检测前处理试剂盒*。2.3 仪器和设备2.3.1 高效液相色谱仪:配紫外-可见光波长检测器。2.3.2 匀浆机。2.3.3 固相萃取机2.3.4 离心机:4000 r/min。2.3.5 调速多用振荡器。2.3.6 聚四氟乙烯离心管: 2.5 mL,50 mL,具塞。2.4 样品制备准确称取已捣碎的样品5.00 g于50 mL离心管中,先加入四环素、沙星类快速检测前处理试剂盒中的提取剂 (液体20mL),用调速多用振荡器150 rpm振荡3 min,,4000 r/min离心5 min,收集上清液10mL加入四环素专用层析柱中(使用前依次用5mL甲醇、5mL提取剂、5mL水激活)挤干,用2mL提取剂洗涤,用0.80mL甲醇洗脱,收集洗脱液,用0.2mL流动相定容至1.0mL。供仪器测定。2.5 测定2.5.1 液相色谱条件a) 色谱柱: C18柱,250 mm×4 mm(i.d.),粒度5µ m b) 流动相: 0.05 mol/L磷酸/三乙胺缓冲液(pH2.4)+乙腈(80+20,V/V) c) 流速: 1.0mL/min d) 柱温: 室温 e) 检测波长: 四环素类紫外检测器350 nm。沙星类 紫外检测器310nm;荧光检测器激发波长280nm,发射波长450nm。f) 进样量:50 uL。3.5.2 标准工作曲线绘制移取各移取四环素类、沙星类混和标准液,用流动相稀释成20 ng/mL、50 ng/mL、250 ng/mL、500 ng/mL标准工作溶液。按液相色谱条件(3.5.1)进行测定,以色谱峰的峰面积为纵坐标,与其对应的浓度为横坐标作图,绘制标准工作曲线,标准工作曲线范围:20.0~500 ng/mL。 3.5.3 试样测定 用微量进样器准确吸取试样溶液(3.4),按液相色谱条件(3.5.1)进行测定,记录色谱峰的保留时间和峰面积。3.6 结果计算按式(1)分别计算供试样品中的四环素类、沙星类残留量。 2×ci×Vω= …… (1)mω-水产品中四环素、沙星类残留量,μg/kg;ci -标准曲线上查出试样溶液中四环素、沙星类标准工作溶液的浓度,(μg/L);V-最终定容体积数,mL;2-换算常数;m-供试试料样品重量,g。本方法分别计算四环素类、沙星类结果。3.7 检测限本方法土霉素、四环素检测限为20µ g/kg;金霉素、强力霉素的检测限为50µ g/kg,沙星类为:5µ g/kg3.8 回收率 本方法土霉素、四环素、金霉素、强力霉素回收率为:75%~85%;沙星类回收率为:75%~85%相关谱图附件可见联 系 人:王 伦 手 机:13810239506 EMAIL:wwj613@sina.com
按国标14931.1做了一个畜禽肉中土霉素四环素金霉素的残留,标准上出峰顺序是土霉素、四环素、金霉素,我做出来的前两个峰没有分离出来,第三个峰出来了,不知原因。 流动相:乙腈/0.01mol/L磷酸二氢钠(用30%硝酸调PH2.5),35:65 柱子:ODS-C18 6.2×15 流速:1.0 波长:355我没有ODS-C18 6.2×15的柱子,用的是C18柱子因为着急认证,请知道的给予帮助。 新手啊 谢谢
0.05mol/L pH0.05的磷酸缓冲液怎么配啊?
请问一下做微生物时所用的接种环是什么样子,是自制还是购的。再就是经初发酵后用接种环将培养物转接到EC培养液中,这里说的培养物是不是初发酵后,试管里面产酸产气的“倒管(小玻璃管)”? 请教啦.....
用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]MS分析四环素类物质,已经在流动相中加了5mmol的草酸,土霉素物质峰型还比较好,四环素有点前沿峰,金霉素和强力霉素拖尾特别严重,前面有好几个小峰中间一直分不开的。有没有分析过相关物质的大神帮忙分析一下,这种情况要怎么解决呢?
1.实验目的 据标准GB/T 14931.1-94测定畜禽肉中土霉素、四环素、金霉素残留量。最低检出浓度为:0.15,0.20,0.65mg/kg。 2.试验原理 样品经提取,微孔滤膜过滤后直接进样,用反相色谱分离,紫外检测器检测,与标准比较定量,出峰顺序为土霉素、四环素、金霉素。标准加法定量。 3.试剂 3.1乙腈(分析纯)。 3.20.01 mol/L磷酸二氢钠溶液:称取1.56 g(±0.01g)磷酸二氢钠(NaH2PH4.2H2O)溶于蒸馏水中,定容到100 mL,经过滤器过滤(选用微孔滤膜为0.45 μm),备用。 3.3土霉素(OTC)标准溶液:称取土霉素0.0100g(±0.0001g),用0.1mol/L盐酸溶液每毫升含土霉素1mg。 3.4四环素(TC)标准溶液:称取四环素0.0100g(±0.001g),用0.01 mol/L盐酸溶液溶解并定容10.00 mL,此溶液每毫升含四环素1mg。 3.5金霉素(CTC)标准溶液:称取金霉素0.0100g(±0.0001g),溶于蒸馏水并定容成10.00mL,此溶液每毫升含金霉素1mg。以上标准品均按1000单位/mg折算。3.3~3.5溶液应于4℃以下保存,可使用1周。 3.6混合标准溶液:取3.3、3.4标准溶液各1.00 mL,取3.5标准溶液2.00 mL,置于10mL容量瓶中,加蒸馏水至刻度。此溶液每毫升含土霉素、四环素各0.1mg,金霉素0.2mg,临时现配。 3.75%高氯酸溶液。 4.仪器 4.1 高效液相色谱仪(HPLC):具紫外检测器。 4.2 振荡器:郑州中谱仪器设备有限公司 4.3 过滤器:郑州中谱仪器设备有限公司 4.4 超声波:郑州中谱仪器设备有限公司 5.色谱条件 5.1柱:ODS-C18 (5 μm) 6.2mm×15 cm。 5.2检测波长:355 nm。 5.3灵敏度:0.002 AUFS。 5.4柱温:室温。 5.5流速:1.0mL/min。 5.6进样量:10 μL。 5.7流动相:乙腈/0.01 mol/L磷酸二氢钠溶液(用30%(V/V)硝酸溶液调节pH2.5),35:65(V/V),使用前用超声波脱气10 min。 6.操作方法 6.1样品测定:称取5.00 g(±0.01 g)切碎的肉样(<5 mm),置于50 mL锥形烧瓶中,加入5%高氯酸25.0mL,于振荡器上振荡提取10 min,移入到离心管中,以2000 r/min离心3min,取上清液经0.45μm滤膜过滤,取溶液10μL进样,记录峰高,从工作曲线上查得含量。 6.2工作曲线:分别称取7份切碎的肉样,每份5.00 g(±0.01 g),分别加入混合标准溶液0、25、50、100、150、200、250 μL(含土霉素、四环素各为0、2.5、5.0、10.0、15.0、20.0、25.0 μg, 含金霉素0、5.0、10.0、20.0、30.0、40.0、50.0 μg), 按6.1方法操作,以峰高为纵坐标,以抗生素含量为横坐标,绘制工作曲线。
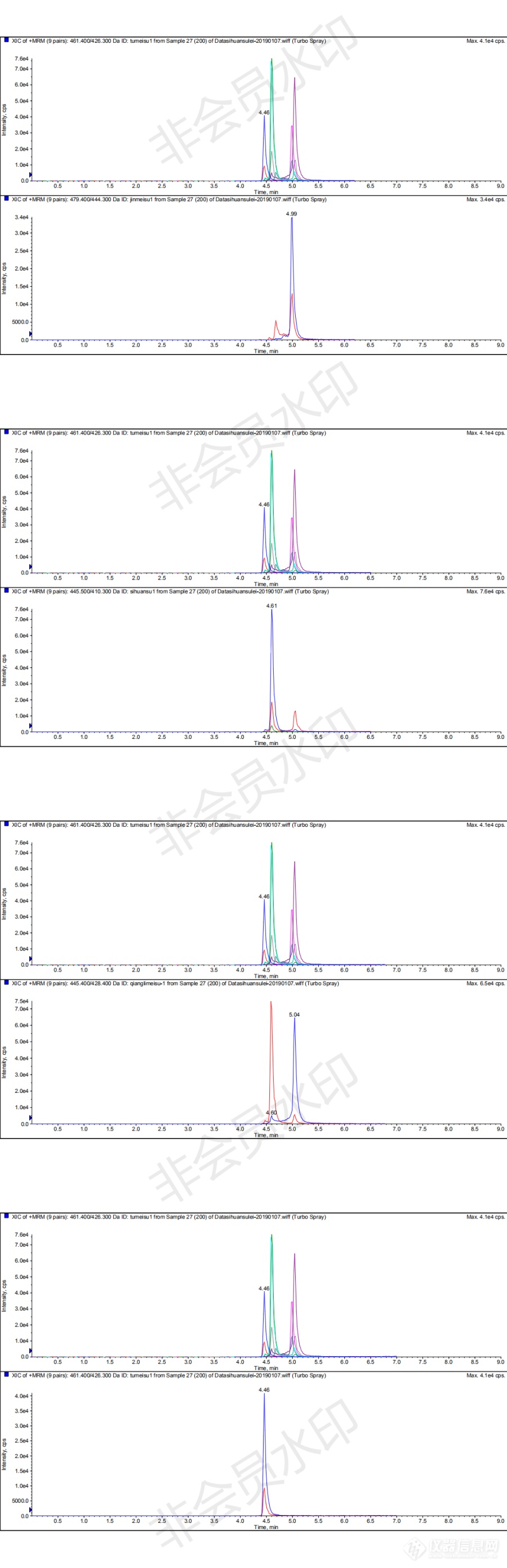
[img=,690,2132]https://ng1.17img.cn/bbsfiles/images/2019/01/201901091446105996_1884_3101087_3.png!w690x2132.jpg[/img][img=,690,2132]https://ng1.17img.cn/bbsfiles/images/2019/01/201901091446105996_1884_3101087_3.png!w690x2132.jpg[/img]这是用AB4000刚走的四环素类4项物质的MRM及各自提取离子图,进的是流动相配制的纯标200ng/mL,四环素和金霉素的储备母液是刚配制的(但并不是刚刚购买的)四环素和金霉素的峰前一直有奇怪的杂峰,强力霉素和四环素好像也没有完全分开[img]https://simg.instrument.com.cn/bbs/images/default/em09511.gif[/img],就土霉素的峰还能看,流动相A:5mmol/L乙酸铵+0.1%甲酸水溶液;B:0.1%甲酸乙腈溶液。有老师能帮忙解释下四环素和金霉素峰形怪异的原因吗,对检测定量有什么影响吗?
大规模动物细胞培养的问题及对策动物细胞培养开始于本世纪初,到1962年规模开始扩大,发展至今已成为生物、医学研究和应用中广泛采用的技术方法,利用动物细胞培养生产具有重要医用价值的酶、生长因子、疫苗和单抗等,已成为医药生物高技术产业的重要部分。由于动物细胞体外培养的生物学特性、相关产品结构的复杂性和质量以及一致性要求,动 物细胞大规模培养技术仍难于满足具有重要医用价值生物制品的规模生产的需求,迫切需要进一步研究和发展细胞培养工艺。目前,世界众多研究领域集中在优化细胞培养环境、改变细胞特性、提高产品的产率并保证其质量和一致性上。1.细胞培养环境细 胞培养环境中抑制因素的积聚是提高细胞密度的主要限制因素。体外动物细胞培养中氨离子的积累是抑制细胞生长的主要因素之一。氨的积聚使细胞内UDP氨基己 糖(UDP-N-乙酰葡糖胺和UDP-N-乙酰半乳糖胺)增加,影响细胞的生长及蛋白的糖基化过程。氨抑制Gln代谢途径,使Asp和Glu消耗增加。细胞消耗Asp增加,可能是细胞线粒体膜上苹果酸-天冬氨酸泵转运NADH加快,使细胞维持糖酵解途径的需要。Asp消耗增加可能会从Gln代谢多经天冬氨 酸转氨酶途径而不是丙氨酸转氨酶或谷氨酸脱氢酶途径得以补偿。氨来源于两方面:一是直接来源于培养基,一是细胞代谢所产生。但两者都涉及谷氨酰胺,因此需要防止培养基中Gln自然分解,限制Gln用量,并尽量去除培养基中的氨。乳酸是细胞糖代谢的产物。高浓度乳酸会抑制乳酸脱氢酶(LDH),从而减少乳酸产生。LDH受抑制后阻止了NADH向NAD的再生及其偶联的丙酮酸/乳酸转 换,从而导致NADH[font=T
做土霉素四环素金霉素,高氯酸溶剂峰特别乱,严重影响出峰。方法是5009.116-能力验证样品是粉末。不用内标行么?用外标做回收率,然后把样品的峰面积按回收率乘回去?样品放一周了,不知道会不会消解。
各位大虾,我用GB/T21317-2007液相方法检测四环素和金霉素,流动相是甲醇,乙腈和10mmol的三氟乙酸,C8柱,但是出了四个峰,做了不同浓度,峰面积和浓度的变化一致,请问大家是否遇到类似问题,如何解决啊
(一)食品中四环素族抗生素药物的限量四环素类抗生素,包括金霉素、土霉素、四环素等,为抑菌性广谱抗生素,除革兰阳性、阴性细菌外,对立克次氏体、衣原体、支原体、螺旋体均有作用,广泛用于多种细菌及立克次氏体、衣原体、支原体等所致的感染。它们自1948年问世以来,陆续被应用于临床,并已成为应用最多、最广泛的广谱抗菌素,目前四环素是我国广泛运用于防治感染性疾病的兽药之一,畜禽喂饲了一定量的四环素后,如代谢时问不足,则残留于肌肉及各器官组织内,食用这些畜禽后,不仅损害人类健康,且由于人体内的病原菌长期接触这些低浓度的药物,从而产生对四环素的耐药性,引起人类和动物感染性疾病治疗的失败。许多国家对TCs残留实施例行监控,欧盟及我国均规定牛乳中四环素类抗生素的最大残留限量为0.1mg/kg。(二)测定土霉素、四环素、金霉素残留的方法①GB/T 20764—2006可食动物肌肉中土霉素、四环素、金霉素、强力霉素残留量的测定。适用于牛肉、羊肉、猪肉、鸡肉和兔肉中土霉素、四环素、金霉素、强力霉素残留量的测定。其方法是用O.1mol/L Na2EDTA—Mcllvaine(pH值为4.00±0.05)缓冲溶液提取可食动物肌肉中的四环素族抗生素残留,提取液经离心后,上清液用Oasis HLB或相当的固相萃取柱和羧酸型阳离子交换柱净化,以乙腈十甲醇+0.01mol/L草酸溶液(2+l+7)为流动相,液相色谱一紫外检测器测定。该法检出限量:土霉素、四环素、金霉素、强力霉素均为O.005mg/kg。②GB/T 18932.23 2003蜂蜜中土霉素、四环素、金霉素、强力霉素残留量的测定方法——液相色谱一串联质谱法,适用于蜂蜜中土霉素、四环素、金霉素、强力霉素残留量的测定。试样中四环素族抗生素残留,用O.1mol/L Na2EDTA—Mcllvaine(pH值为4.OO±0.05)缓冲溶液提取,提取液经离心后,上清液用Oasis HLB或相当的固相萃取柱和阴离子交换柱净化,采用配有电喷雾离子源的液相色谱一串联四极杆质谱仪测定。该法检出限:土霉素、四环素为0.01mg/kg;金霉素、强力霉素为0.02mg/kg。③DB33T 691—2008水产品中土霉素、四环素、金霉素、强力霉素残留量的测定——高效液相色谱荧光检测法,适用于水产品中土霉素、四环素、金霉素、强力霉素残留量的测定:样品经柠檬酸缓冲液提取,正己烷脱脂,Oasis HLB固相萃取柱净化,以咪唑缓冲溶液一甲醇为流动相,用高效液相色谱荧光检测器检测,外标法定量。本方法检出限为土霉素0.0lmg/kg,四环素0.01mg/kg,金霉素O.03mg/kg,强力霉素0.03mg/kg。
中国环科院培训资料 大气+固废方面[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=177755]中国环科院培训资料 大气+固废方面.rar[/url]
亲们,求助一下,用GB/T 20764-2006 《可食动物肌肉中土霉素、四环素、金霉素、强力霉素残留量的测定 液相色谱-紫外检测法》做抗生素,仪器:WATERS e2695,检测器:2698 DAD,柱子:Sunfire C18 250*4.6,5,流动相:乙腈+甲醇+0.01mol/L草酸溶液(2:1:7),流速:1.0mL/min,柱温:30度,检测波长:350nm,进样量:20微升,用标曲最大点进样(200ng/mL),四种都不出峰,我们试了更大浓度的也一样。请问问题出在那里呢?有推荐的方法和柱子吗?
哪位做过禽肉中金霉素、土霉素、四环素测定啊?出峰好慢,流动相用:乙腈+0.01mol/l磷酸二氢钠(30%硝酸调节PH2.5)=35+65.40多分钟了还没出峰,流速2.0ml/min ,C18的柱子
0.05mol/L PH5.0的磷酸-柠檬酸缓冲液怎么配,PH我知道怎么调,但是浓度是怎么确定的?
请问岛津的贝斯培压环是什么材料?

[align=center][b]这次培训,收获满满[/b][/align][align=center][b] ----记环评检测技术培训[/b][/align][align=center] [/align] 为了提高检测水平,优化工作效能,4月15日-6月11日,中心组织安排了无公害农产品产地环境检测技术培训,通过近两个月的学习,使我收获满满,受益匪浅,总结几点收获和感触记下与大家共勉。一、培训内容这次培训的形式是理论知识讲解与实际操作相结合,主要包括无公害农产品环评检测项目,有效数字的修约和计算,检出限和相对偏差,检测数据原始记录的填写,自己所分参数练习,仪器操作,加标试验等,最后进行了考试总结。[align=center][img=,450,448]https://ng1.17img.cn/bbsfiles/images/2019/07/201907081709319174_4560_3424753_3.jpg!w450x448.jpg[/img][/align][align=center][/align][align=center][/align][align=center][/align][align=center][/align][align=center][/align][align=left]二、培训感受与收获[/align][align=center][/align][align=left]1.领导重视,授课老师专业敬业。[/align][align=center][/align][align=left][color=#191919]这次培训,我感到机会来之不易,中心领导提前对培训进行计划和安排,宋老师精心编写了培训教材,讲课的宋老师学识渊博、经验丰富,并结合日常工作实际,以问题为导向,找准学员短板,针对性有侧重地开展教学,做到有的放矢,将一些我们平素认为空洞的理论讲得栩栩如生,使一些吃的不透的理论在这里得到了升华,让一些以前不能理解的现实问题在这里被“冲击”得透彻分明,既有理论的高度,又有很强的现实针对性、可操作性。[/color][/align][align=center][b][/b][img=,447,451]https://ng1.17img.cn/bbsfiles/images/2019/07/201907081708526644_7199_3424753_3.jpg!w447x451.jpg[/img][/align][align=left]2.同事们学习热情高涨,互相探讨,共同进步。[/align][align=center][/align][align=left]这次参加培训的都是从事检测的人员,都是我的良师益友,他们各有所长,更让我体会到自己的不足。[color=#191919]在和谐活泼的气氛中共同探讨、互相切磋、集思广益,学他人之长、见贤思齐,还出现许多有创新、有深度的思想观点,让我沉浸在品味“营养大餐”的环境中,既学到了知识,又交流了思想。我感受到我们检测人员不仅有严谨细致的一面,也有活泼可爱的一面。[/color][/align][align=center][/align][align=left][color=#191919]3.[/color][color=#191919]业务素质得到了提升[/color][/align][align=center][/align][align=left]首先,重实用,理论与实操相结合。此次培训,根据日常工作需要,讲了常用的理论知识,然后参数练习,加标测试,上机操作,现场操作,现场指导,规范了实验室操作动作,出现检验数据不当时,如空白偏高,加标回收率低等问题时,又反复试验,查找各种原因,[color=#191919]并以理论结合实际,[/color]以理论指导实际,在工作中学习,在实践中领悟。[/align][align=center][/align][align=left]例 如1:检出限的测定,一般将3倍空白值的标准偏差(测定次数n20)相对应的质量或浓度称为检出限,即L=3S/b。先按照分析方法的步骤,设置6个以上标准系列浓度点,各浓度点的测量信号值减去零浓度点的测量信号值,经回归方程计算后绘制校正曲线,校准曲线的相关系数接近或达到0.999,小于时,应对校准曲线各点测定值进行检验或重新测定,当接近或达到0.999时即符合要求。校准曲线不合格不能使用,使用时,不得随意超出标准系列浓度范围,不得长期使用,然后再做20个空白值测定,按照上述公式计算检出限。通过这次理论学习和实际操作,使我熟练掌握了检出限这个概念,并能熟练计算。[/align][align=center][/align][align=center][img=,553,641]https://ng1.17img.cn/bbsfiles/images/2019/07/201907081715115304_6700_3424753_3.jpg!w553x641.jpg[/img][/align][align=center][/align][align=center][/align][align=center][/align][align=left]例如2:我学习了均值与相对偏差的计算,并能用数字修约的规定计算出正确的结果。[/align][align=center][/align][align=left]1.均值的计算:例如编号HJ2012W001-S-01的样品测定结果一栏中两次结果分别为0.0011、0.0014,那么均值=[sub][img=,109,41]https://ng1.17img.cn/bbsfiles/images/2019/07/201907081711023416_3310_3424753_3.gif!w109x41.jpg[/img][/sub]=[sub][img=,51,41]https://ng1.17img.cn/bbsfiles/images/2019/07/201907081711150937_2387_3424753_3.gif!w51x41.jpg[/img][/sub]=0.00125,因为[sub][/sub]中[img=,51,41]https://ng1.17img.cn/bbsfiles/images/2019/07/201907081711150937_2387_3424753_3.gif!w51x41.jpg[/img]0.0025是2位有效数字,2是自然数,所以计算结果应该保留2位有效数字,0.00125保留两位有效数字为0.0012。 [/align][align=left][/align][align=left]2.相对偏差的计算:相对偏差(%)=[img=,44,41]https://ng1.17img.cn/bbsfiles/images/2019/07/201907081712437320_8794_3424753_3.gif!w44x41.jpg[/img]*100%,例如编号HJ2012W001-S-01的两次平行测定结果分别为0.0011、0.0014,相对偏差的计算是:相对偏差(%)= [sub][img=,109,41]https://ng1.17img.cn/bbsfiles/images/2019/07/201907081712521214_9504_3424753_3.gif!w109x41.jpg[/img][/sub]*100= [sub][img=,51,41]https://ng1.17img.cn/bbsfiles/images/2019/07/201907081713085131_2905_3424753_3.gif!w51x41.jpg[/img][/sub] *100=0.12*100=12%,因为[img=,51,41]https://ng1.17img.cn/bbsfiles/images/2019/07/201907081713085131_2905_3424753_3.gif!w51x41.jpg[/img] *100中0.0003是一位有效数字,所以计算结果应保留一位有效数字,但12%是两位有效数字,必须把两位变成一位,应该保留成1*10%。通过培训,心里清楚了,再填写原始记录时,很容易就能正确填写了。[/align][align=center][/align][align=center][img=,554,483]https://ng1.17img.cn/bbsfiles/images/2019/07/201907081714286755_7091_3424753_3.jpg!w554x483.jpg[/img][/align][align=center][/align][align=center][/align][align=center][/align][align=center][/align][align=left][color=#191919]其次,重实效,学习与测试相结合。通过测试,让我真真切切认识到了自己的短板与不足,也让讲课老师更有针对地进行讲课,有侧重地开展教学,靶向补短。这种以考促学求实训,以学促考强本领的培训方式,虽然让我感到有压力,但也给我增添了学习动力。经过考试,与其他同事交流,讲课老师分析讲解,发现了不足,对自身业务知识有了新的认识,更需要加强学习。[/color][/align][align=center][/align][align=center][img=,449,448]https://ng1.17img.cn/bbsfiles/images/2019/07/201907081716209633_2492_3424753_3.jpg!w449x448.jpg[/img][/align][align=center][/align][align=left][color=#191919]这次培训学习,[/color]使我收获满满,受益匪浅。更让我深刻认识到,要更快的提高检测水平,[color=#191919]必须坚持学习,勤于思考,不断充实自己。根据客观实际,在认真学习、借鉴的基础上,灵活运用所学的知识和积累的经验,要对标问题,补齐短板,积极培养埋头苦干、顽强拼搏、勇于奉献的精神,努力做到学以致用,练就做好检验工作的真本领,争做一名合格的检验工作者。 [/color][/align][align=left][/align][align=center][color=#191919] [/color][/align][align=center][/align][align=center][/align][align=center][/align][align=center][/align][align=center][/align][align=center][/align][align=center][/align][align=center][/align][align=center][/align][align=center][/align]
最近在用高液做四环素类;我参考文献用乙腈:甲醇:0.01mol/l的草酸水溶液=2:1:7 等度洗脱,金霉素和多西环素出峰响应值很低而且不成一个尖锐的峰型,拖尾。我加的标准品是4ug/ml的浓度。想参考一下你们使用的条件
实验室液相菜鸟,准备做四环素土霉素金霉素的检测,但是标准一直跑不出来,液相条件摸索不出来,请问有没有大神可以跟随指导,成功的话有偿^ ^帮帮孩子







 我要推广仪器
我要推广仪器
 下载APP
下载APP