循环伏安法 制得的高分子薄膜侧基咔唑作为交联点生成二聚咔唑,想测其反应的咔唑基团数占起始的咔唑基团数的比值谢谢大家,用红外的方法可以么?怎么用?O(∩_∩)O谢谢
谁知道哪里卖知道确切气相色谱含量的咔唑样品啊,只要含量达到九十以上就行啊,知道的朋友请回答啊,谢谢啊。
(一)食品中果胶的意义果胶产品可分为高脂果胶和低脂果胶,酯化度大于50%的称为高脂果胶,酯化度低于50%的称为低脂果胶,果胶的显著特性是有胶凝性和增稠稳定性,并且有很强的耐酸、耐高温性能。果胶在食品工业中应用较广,如利用果胶水溶液在适当条件下可以形成凝胶的特性,生产果酱、果冻及高级糖果等食品;利用果胶具有增稠、稳定、乳化等功能,可以解决饮料的分层、防止沉淀的问题,还可以改善风味等。测定果胶物质的方法有称量法、咔唑比色法、果胶酸钙滴定法、蒸馏滴定法等。(二)咔唑比色法果胶经水解可生成半乳糖醛酸,半乳糖醛酸在强酸中可与咔唑试剂发生缩合反应,生成紫红色化合物,该紫红色化合物的呈色强度与半乳糖醛酸含量成正比,故可通过测定吸光值对果胶含量进行定量。此法适用于各类食品的果胶含量的测定,具有操作简便、快速、准确度高、重现性好等特点。1.样品处理同重量法。2.果胶处理同重量法。3.标准曲线制作取8支50mL比色管,各加入12mL浓硫酸,于冰水浴中边冷却边缓缓依次加入浓度为0、10μg/mL、20μg/mL、30μ/ml、40μ/mL、50μg/mL、60μg/mL、70μg/ml的半乳糖醛酸标准溶液2mL,充分混合后,再置于冰水浴中冷却。然后在沸水浴中准确加热10min,迅速冷却到室温,各加入0.15%咔唑试剂1mL。充分混合,室温下放置30min,以半乳糖醛酸含量为0的半乳糖醛酸标准溶液为空白,在530nm波长下测定吸光值,以半乳糖醛酸含量为纵坐标,吸光值为横坐标,绘制标准曲线。4.样品提取液的测定取果胶提取液,用水稀释到适当浓度(含半乳糖醛酸10~70μg/mL)。取2mL稀释液于50mL比色管中,以下按制作标准曲线的方法操作,测定吸光值。从标准曲线上查出半乳糖醛酸的浓度(μg/mL)。5.结果计算X(以半乳糖醛酸计)=c*V*K/m*106*100式中 X——样品中果胶的质量分数,%;c——从标准曲线上查得的半乳糖醛酸的浓度,肚g/mL;V——果胶提取液的总体积,mL;K——提取液稀释倍数;m——样品质量,g。6.试剂①乙醇。②乙醚。③0.05mol/L盐酸溶液。④O.15%咔唑乙醇溶液:化学纯咔唑0.150g,溶解于精制乙醇中并定容到100mL,咔唑溶解缓慢,需加以搅拌。精制乙醇:取无水乙醇或95%乙醇1000mL,加入锌粉4g,(1+1)硫酸4mL,在水浴中回流10h,用全玻璃仪器蒸馏,馏出液每1000mL加锌粉和氢氧化钾各4g,重新蒸馏一次。⑤半乳糖醛酸标准溶液:半乳糖醛酸100mg,溶于蒸馏水并定容到100mL,用此液配制一组浓度为10~70μg/mL的半乳糖醛酸标准溶液。⑥硫酸。7.仪器①分光光度计。②经50mL比色管。8.注意事项①本法的测定结果以半乳糖醛酸表示,不同来源的果胶中半乳糖醛酸的含量不同,如甜橙为77.7%,柠檬为94.2%,柑橘为96%,苹果为72%~75%,若把结果换算为果胶的含量,可按上述关系计算换算系数。②样品处理时应充分洗涤去除糖分,减少其存在对咔唑的呈色反应的影响。③在测定样液和制作样液标准曲线时,应使用相同规格、同批号的浓硫酸,以保证浓度一致,减少硫酸浓度对咔唑的呈色反应的影响。
【关键词】资料分享 可以有 做广告 不许有 ——土豆批改 内容摘要:果胶经水解生成半乳糖醛酸,在硫酸中与咔唑试剂发生缩合反应,生成紫红色化合物,其呈色强度与半乳糖醛酸含量成正比,可比色定量。 (中检所标准物质) 咔唑比色法 此法适用于各类食品,且操作简便、快速,准确度高. 1.原理 果胶经水解生成半乳糖醛酸,在硫酸中与咔唑试剂发生缩合反应,生成紫红色化合物,其呈色强度与半乳糖醛酸含量成正比,可比色定量。 2.仪器 ①分光光度计。 ②50 mI,比色管。 3.试剂 ①99%乙醇。 ②70%乙醇. ③yi醚。 ④0.05 tool·L叫盐酸溶液。 ⑤0.5。tool·L叫氢氧化钠。 ⑥O.15%咔唑乙醇溶液:称取化学纯咔唑O.15 g,溶解于精制乙醇中并定容到1。0 mI咔唑溶解缓慢,需加以搅拌。 ⑦精制乙醇:取无水乙酵或95%乙醇1 000 mL,加入锌粉4 g,硫酸(1:1)4 mI_,,在水浴中回流10 h,用全玻璃仪器蒸馏,馏出液每1 000 mI.加锌粉和氢氧化钾各4 g,重新蒸馏一次。(药检所对照品) ⑧半乳糖醛酸标准贮备溶液:准确称取半乳糖醛酸100 mg,溶于蒸馏水并定容到100mL,得浓度为l mg·mI.1半乳糖醛酸标准贮备液。 ⑨半乳糖醛酸标准工作液:分别准确吸取0.0、1.0、2.O、3.O、4.O、5.O、6.O、7.0 ml。半乳糖醛酸标准贮备溶液于8个10¨。mL容量瓶中,用水稀释至刻度,得一组浓度分别为0.0、10、20、30、40、50、60、70扯g·mI。叫的半乳糖醛酸标准工作液。 ⑩浓硫酸:优级纯。 4.测定步骤 ①提取果胶 a.水溶性果胶提取:用150 mI,水将上述挥发至干的残渣移人250 mI.烧杯中,加热至沸并保持沸腾1 h,随时补足蒸发的水分,冷却后移人250 mI。容量瓶中,加水定容,摇匀,过滤,弃去初滤液,收集滤液即为水溶性果胶提取液。 b.总果胶的提取:用150 mL加热至沸的0.05 tool·L叫盐酸溶液把挥干的残渣移人250mL锥形瓶中,装上冷凝器,于沸水浴中加热回流1 h,冷却后移人250 ml。容量瓶中,加甲基红指示剂2滴,加O.5 mol·L1氢氧化钠中和后,用水定容,摇匀,过滤,收集滤液即为总果胶提取液。(北京标物中心) ②样品处理 a.新鲜样品:称取试样30~50 g,用小刀切成薄片,置于预先放有99%乙醇的500 mI。锥形瓶中。装上回流冷凝器,在水浴上沸腾回流15 rain后,冷却。用布氏漏斗过滤,残渣移至研钵中,一边慢慢磨碎,一边滴加70%的热乙醇,冷却后再过滤,反复操作至滤液不呈糖的反应(用苯酚一硫酸法检验,见说明)为止。残渣用99%乙醇洗涤脱水,再用yi醚洗涤以除去脂类和色素,漏斗中残渣在空气中挥发去yi醚。 b.干燥样品:研细,过60目筛,称取5~10 g样品于烧杯中,加入热的70%乙醇,充分搅拌以提取糖类,过滤。反复操作至滤液不呈糖的反应。残渣用99%乙醇洗涤,再用yi醚洗涤,最后让yi醚挥发掉。 ③标准工作曲线的制作:取8支50 mL比色管,各加入12 mI。浓硫酸,置冰水浴中,边冷却边缓缓依次加入浓度为O.0、10、20、30、40、50、60、70肚g·ml,叫的半乳糖醛酸标准溶液2mL,充分混合后,再置冰水浴中冷却。然后在沸水浴中准确加热10 min,用流动水迅速冷却到室温,各加人O.15%咔唑试剂1 mI。,充分混合,置室温下放置30 rain,以O号管为空白在530nm波长下测定吸光度,绘制标准工作曲线。(中华标准物质网) ④测定:取果胶提取液,用水稀释到适当浓度(含半乳糖醛酸10~70肛g·mLl)。取2ml。稀释液于50 mI.比色管中,以下按制作标准曲线的方法操作,测定吸光度。从标准曲线上查出半乳糖醛酸浓度(ug·mI。) 5.计算 6.说明 ①糖分的存在对咔唑的呈色反应影响较大,使结果偏高,故样品处理时应充分洗涤以除去糖分。 ②检验糖分的苯酚一硫酸法:取检液l mL,置于试管中,加入5%苯酚水溶液1 ml。,再加入硫酸5 mL,混匀,如溶液呈褐色,证明检液中含有糖分。 ③硫酸浓度对呈色反应影响较大,故在测定样液和制作标准曲线时,应使用同规格、同批号的浓硫酸,以保证其浓度一致。土豆:感谢分享资料,但是含有广告内容是不允许的哦,亲,下次别违规了。
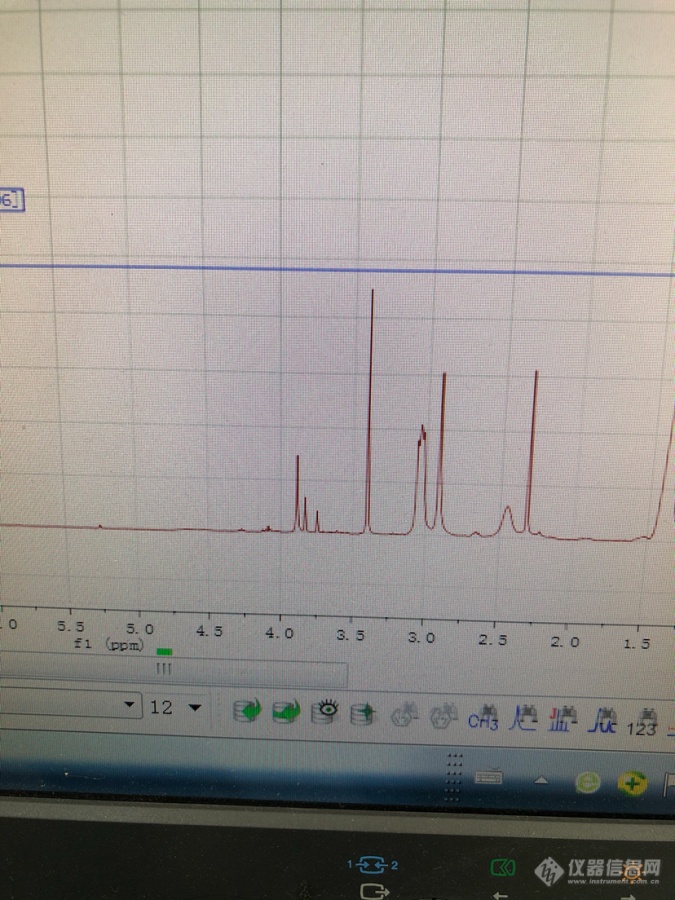
[color=#444444]做的是聚(2,7-咔唑)衍生物 [/color][color=#444444]甲苯作溶剂 反应后萃取浓缩[/color][color=#444444]可是滴入甲醇中 无沉淀 最后都是粘稠的液体 无固体沉出[/color][color=#444444]所以反映结束后处理该怎么做呢 多次进行溶解沉出 都是沉不出固体[/color][color=#444444]测核磁3-4的峰 可能是甲苯和甲醇未干吗?[/color][color=#444444][img=,675,900]https://ng1.17img.cn/bbsfiles/images/2019/05/201905151636301368_4040_1646718_3.png!w675x900.jpg[/img][/color]
苄基频哪醇硼酯与硼相连的苄位碳在碳谱中似乎扫不出来,为什么?
6苄基腺嘌呤为什么常温保存?望老师不吝赐教
[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]做116种组分,萘,六氯丁二烯线性都没啥问题,唯独苄基氯线性不太好。不知道哪位大佬把苄基氯做好了,分享一下经验或者需要注意的细节。
[color=#444444]求问ESI-ms是否可能把 苄基作为保护基的糖的改造产物打成碎片~[/color][color=#444444]今天所得到的质谱峰非常明显(1163和455,相对丰度是100和15),但不是目标产物的分子量。但是这两个峰的m/z相加所得是目标分子量~是否存在这种可能[/color]
做6-苄基腺嘌呤方法验证的时候色谱柱一直平衡不好,流动相用的是甲醇+0.02mol/L乙酸铵(千分之1乙酸),基线一直是这样的,有什么解决办法吗?[img]https://ng1.17img.cn/bbsfiles/images/2021/05/202105181351571722_3426_5026084_3.png[/img]
各位大侠好,有谁能提供苄基三甲基氯化铵分析方法吗?
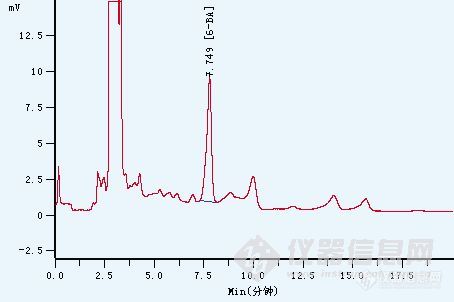
分享一个我公司实验人员优化的豆芽中的6-苄基腺嘌呤检测方法。豆芽中的6-苄基腺嘌呤检测,按照DB/11T 279-2006,北京市地方检测方法,用C18小柱富集,回收率只有40%左右,考虑到6-苄基腺嘌呤是酸性的物质,后来尝试了用CNW poly-sery MAX小柱富集,回收率可以做到80%以上。前处理方法:捣碎豆芽10g用20mL 酸化甲醇(甲醇/水=60:40,加入50μL乙酸)提取,离心,上清液旋转蒸发除去甲醇,调pH至9.0左右,待上样至固相萃取小柱。SPE固相萃取:活化:5mL 甲醇平衡:5mL 去离子水上样:流速 0.5滴/s淋洗:3mL 去离子水,3mL 20%甲醇水溶液,抽干洗脱:5mL 2%乙酸甲醇液相色谱条件:检测波长(nm): UV267nm柱型号: C18柱(4.6*250,5μm)流量(ml/min): 1.0mL/min流动相: 甲醇/1%乙酸=50/50柱温(℃): 30进样量: 20μL附上图谱(3ppm目标物)http://ng1.17img.cn/bbsfiles/images/2011/09/201109301339_320394_1776417_3.jpg
2011年9月7日消息,法国健康产品安全机构(AFSSAPS)宣布一项紧急禁令,要求生产商禁止在化妆品中使用3-亚苄基樟脑(3-Benzylidene-camphor,3-BC),原因是该物质具有潜在风险会干扰人体内分泌。非政府组织国际化学秘书处(ChemSec)对该决定表示欢迎,并呼吁法国按照REACH法规将3-亚苄基樟脑拟定为高度关注物质(SVHC)。ChemSec 称,3-亚苄基樟脑常作为紫外线过滤层在防晒剂中使用,该物质可通过皮肤被人体吸收,目前已在孕妇母乳中发现3-BC的存在。AFSSAPS也在决议声明中表示,最近的研究表明3-BC会干扰内分泌,特别是影响生育问题,含有该物质的化妆品可能对人类的健康造成严重的危害。因此,机构决定即刻引进禁令,禁止在化妆产品中使用该物质。生产商、进口商,以及分销商必须尽快采取措施停止将产品投放市场并将未销售的产品从货柜上撤离。ChemSec项目协调员对法国颁布3-BC禁令的举动表示支持,并希望能将禁令扩展至欧盟范围,鼓励法国及欧盟其他成员国仿效德国,将内分泌干扰物质增加至REACH候选名单中。
请问大家有没有对"N-苄基马来酰亚胺"这个物质做过分析的?能不能告诉我它的分析方法是什么?条件是什么样的?谢谢![em24]
dongxianwenbianan为苄胺的红外光谱,dongxianwen20应该是苄基马来酰亚胺的光谱,怎么分析3000以上的峰?请教各位老师,如何分析酰胺中的C—N和稀烃的C—H?
求购二苄基二硫代乙二酰胺,分析纯,25g/瓶或50g/瓶,单价,和联系方式电话:13550052124 E:Email:wangke_wj@126.com
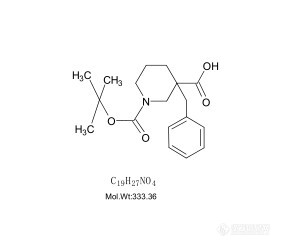
色谱世界的各位大侠们,1-boc-3-苄基哌啶甲酸文献报道说用4.6mm*150mm C18的二氧化硅柱子,用乙腈、水、三氟乙酸作为流动相,在214和254处有吸收峰。我们用紫外全波长扫描后,发现只有在204处有紫外吸收峰,可是我们用乙腈和水作流动相,这个东西在214处不出峰。这个东西该怎么检测纯度呢。这个化合物是通过1-boc-3-哌啶甲酸 和溴苄合成的, 还有1-boc-3-哌啶甲酸 的熔点是159-162℃。 我们这个东西的熔点是109-116℃。 所以这两个东西很定有是不一样的。 实在不行只有打核磁了。我们这个原料的紫外吸收也在204这个位置。但我们用254的紫外薄层检测,发现原料不显色,1-boc-3-苄基哌啶甲酸 轻微显色。[img=,281,247]https://ng1.17img.cn/bbsfiles/images/2019/07/201907231115067664_5937_1815404_3.jpg!w281x247.jpg[/img]
来源:科学网 作者:马 臻正在工作中,突然收到一个读者心急如焚的电话:“马老师,我给一个杂志投了篇稿子,一个星期过去了,还没有送审,要不要催编辑?”我听了,见怪不怪!编辑收到稿件,总要看了再送审。一个星期算什么,一个月都不稀奇。催编辑不是错,但是错就错在不能把握好自己的语言。曾有人心急如焚地把他给编辑发的信给我看,说是编辑看了这几封连续的催稿信,不但把目前的稿子枪毙了,把已经接收的稿子都撤销了。我看了那些充满情绪化语言的催稿信,的确很恼怒。急着要毕业,这不是你的错,但是以需要文章毕业为由催编辑,这在国外看来是不可理喻的:你要发文章毕业,那你为什么不能早点投呢?你抱怨这个杂志处理速度慢,那你为什么不投审稿快的高档次杂志呢?很多事情都是这样,有些语言没有把握好,会出麻烦。比如有的报刊杂志文章的作者给报刊杂志投稿后,老催问什么时候能刊登,并且常给报社打电话、写电子邮件催稿费。编辑心里就会想你是为了钱而写稿子的。这下可好,以后都不大用这位作者的稿件了。再比如,申请国外的博士后,想得到更高的薪水。但是,自己需要养活全家,这不应该是争取更高薪水的理由:谁叫你把全家带到美国了?既然你决定把全家带到美国,就需要有自己的承担。要是抱怨薪水只够养活乞丐,对方就更生气了。这么说,也不是说不能催编辑。完全能够催编辑,关键在于什么时间,以何种语言催编辑。我曾有篇稿子校对清样以后两个月都没有在线刊登,而该杂志社别的稿子很快在线了,我便写了封信:Dear Editor, Thank you very much for accepting our paper entitled "Co3O4-KIT-6 Composite Catalysts: Synthesis, Characterization, and Application in Catalytic Decomposition of N2O" (MS Number: NANO4808). We feel quite happy publishing with you. We received our proof (proof number: 874) and sent the corrections out two months ago, and we still didn't see its publication online. Could you please kindly check the status of its publication? We do appreciate your kind help. Zhen Ma得到的回复为:Dear Dr. Zhen Ma,Thank you for your e-mail.Your article is being processed in the production and it will be published online within three days.Best regards,Devi
国家重申生产者不得在豆芽生产过程中使用6-苄基腺嘌呤、4-氯苯氧乙酸钠、赤霉素等物质,豆芽经营者不得经营含有6-苄基腺嘌呤、4-氯苯氧乙酸钠、赤霉素等物质的豆芽。大家有豆芽中6-苄基腺嘌呤、4-氯苯氧乙酸钠、赤霉素同时检测方法吗?
最近在做苄基二甲胺(N,N-二甲基苄胺)含量分析的时候,要用到甲酸——乙酸酐混合酰化剂,但方法上没有阐明甲酸——乙酸酐混合酰化剂的具体配制方法,不知两者以什么体积比混合能达到一个最好的酰化效果,有做过这个项目的大虾请指教,谢谢!
2011年9月7日消息,法国健康产品安全机构(AFSSAPS)宣布一项紧急禁令,要求生产商禁止在化妆品中使用3-亚苄基樟脑(3-Benzylidene-camphor,3-BC),原因是该物质具有潜在风险会干扰人体内分泌。非政府组织国际化学秘书处(ChemSec)对该决定表示欢迎,并呼吁法国按照REACH法规将3-亚苄基樟脑拟定为高度关注物质(SVHC)。 ChemSec称,3-亚苄基樟脑常作为紫外线过滤层在防晒剂中使用,该物质可通过皮肤被人体吸收,目前已在孕妇母乳中发现3-BC的存在。AFSSAPS也在决议声明中表示,最近的研究表明3-BC会干扰内分泌,特别是影响生育问题,含有该物质的化妆品可能对人类的健康造成严重的危害。因此,机构决定即刻引进禁令,禁止在化妆产品中使用该物质。生产商、进口商,以及分销商必须尽快采取措施停止将产品投放市场并将未销售的产品从货柜上撤离。 ChemSec项目协调员对法国颁布3-BC禁令的举动表示支持,并希望能将禁令扩展至欧盟范围,鼓励法国及欧盟其他成员国仿效德国,将内分泌干扰物质增加至REACH候选名单中。
最近在做一个糖苷类的检测方法,想用对硝基苯甲酸和糖苷上的羟基发生酯化反应显色后用紫外检测器检测,但是副产物太多,不能进行准确的定量,想请教下还有别的衍生化条件没? 我的衍生化条件是10mg糖苷样品+50mg对硝基苯甲酸加丙酮溶解后加1ml浓盐酸加热10min,条件是我自己摸索的,可能有很多的不规范的地方样品里可能有葡萄糖,蔗糖,葡萄糖甲苷,1-O-甲基-四苄基葡萄糖,1-羟基-四苄基葡萄糖
用乙醚做溶剂,氯化苄和镁条做原料,合成出来的格氏试剂颜色为乳白色絮状物,不知是不是目标产物(苄基氯化镁),特求助同仁!
6-苄基腺嘌呤在被豆芽吸收后会转化成什么物质?有没有对这个植物生长激素有研究的版友指导一下?我的困惑其实是作为检测工作来说,6-苄基腺嘌呤在豆芽样品中残留量会不会随着时间的推移,检出量会不会发生变化?
联苯苄基氯(学名;4,4’-双氯甲基联苯)溶解方法?用ICP-AES测定其中的Ca,Fe,Zn,P,Na等元素(1--5ppm)。请高手指点样品处理方法为盼。谢谢啊http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif
各位老师好:我用的是热电acess max,每天跑完样品时都是半夜了。想编辑一个检测结束后,维护仪器的仪器方法,请问应该编辑哪些参数,怎么编辑?怎么使用?
帖子无法编辑成已应助,编辑后跳转至如下文字,其他帖子都能编辑的http://bbs.instrument.com.cn/shtml/20110509/3295181/错误您所请求的网址(URL)无法获取
化妆品中亚苄基樟脑磺酸检测方法
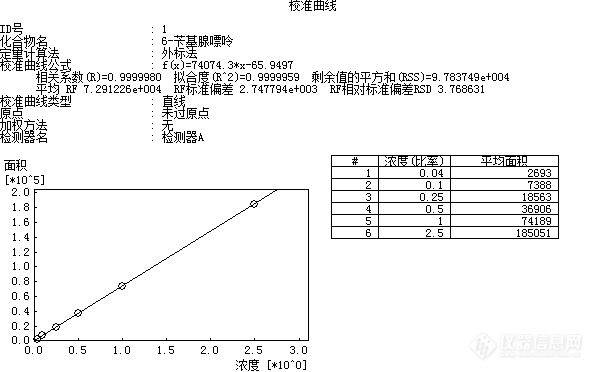
[align=center][b]食品中6-苄基腺嘌呤的测定方法验证报告[/b][/align][align=center][b]GB/T 23381-2009( 高效液相色谱法)[/b][/align][align=center][b]张霞[/b][/align]一、方法概述1.范围 本标准规定了用高效液相色谱法测定食品中6-苄基腺嘌呤(6-BA)含量的方法。 本标准适用于果蔬菜(豆芽、黄瓜、番茄、香菇、草莓、橙类)等植物性食品及其制品中6-苄基腺嘌呤的测定。2.规范性引用文件下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方面研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。GB/T 6682 分析实验室用水规格和试验方法(GB/T 6682-2008,ISO 3696:1987,MOD)3.方法提要 试样经甲醇提取、浓缩并净化后,用高效液相色谱检测,外标法定量。二、仪器与试剂1. 仪器1.1高效液相色谱仪:配有紫外检测器或二极管阵列检测器。1.2 组织捣碎机。1.3离心机:转速不低于4000r/min。1.4超声波清洗仪。1.5旋转蒸发仪。1.6固相萃取装置。1.7电子天平:感量0.1mg。1.8微孔滤膜:0.45μm,有机相。以上仪器符合国标要求。2. 试剂及其配制 除另有规定外,所有试剂均为分析纯,水为GB/T 6682规定的一级水。2.1甲醇:色谱纯。2.2冰乙酸。2.3 C[sub]18[/sub]固相萃取柱:6mL,500mg,或相当者,使用前依次用5mL甲醇、10mL水活化。2.4乙酸铵溶液(0.02mol/L):称取1.54g乙酸铵,用适量水溶解,加入1.0mL冰乙酸,加水定容至1000mL。2.5 6-苄基腺嘌呤标准溶液(100.0μg/mL) [color=#ff0000] [/color][color=#ff0000]来源[/color][color=#ff0000]:[/color][color=#ff0000]农业部环境保护科研监测所[/color][color=#ff0000] [/color][color=#ff0000]货[/color][color=#ff0000]号[/color][color=#ff0000]:[/color][color=#ff0000]SB05-368-2016[/color][color=#ff0000] [/color]三、分析步骤1、标准曲线绘制1.1 标准工作液的配制: 分别吸取适量6-苄基腺嘌呤标准溶液,用甲醇定容至10mL容量瓶中,配制成浓度为0.04μg/mL、0.1μg/mL、0.25μg/mL、0.5μg/mL、1.0μg/mL、2.5μg/mL系列工作液。2、样品的处理2.1提取:称取经组织捣碎机捣碎的样品约10g(精确到0.01g)于50mL离心管中,加入20mL甲醇,超声提取15min,以转速不低于4000r/min离心10min,上清液转入50mL梨形瓶中,样品再次用20mL甲醇超声提取15min,离心合并上清液,用旋转蒸发仪(不超过60℃)浓缩至近干,去除甲醇,残液待净化。2.2纯化:将上述2.1残液以2mL/min流速通过预先活化的固相萃取柱,用少量水(约2mL)洗涤梨形瓶,洗液过固相萃取柱,再用5mL水洗涤固相萃取柱,去除杂质后用甲醇洗脱并定容至5.0mL,混匀后经0.45μm滤膜过滤,作为待测液供HPLC分析。3.仪器测定条件3.1色谱柱:C18柱,柱长250mm,内径4.6mm,粒径5μm或相当型号色谱柱。3.2流速:1.0mL/min。3.3柱温:30℃。3.4检测波长:267nm。3.5进样量:10μL。3.6流动相:甲醇 :0.02mol/L乙酸铵溶液=1:1四、结果处理试样6-苄基腺嘌呤含量按下式进行计算:[table][tr][td=1,2][align=center]X(mg/kg)=[/align][/td][td]C×[i]V[/i]×1000[/td][/tr][tr][td]m×1000[/td][/tr][/table]式中:X-试样中6-苄基腺嘌呤含量,单位为毫克每千克(mg/kg) C-由标准曲线计算出样液中6-苄基腺嘌呤的浓度,单位为微克每毫升(μg/mL) m-试样质量,单位为克(g) V-试样的最终定容体积,单位为毫升(mL)。1000—换算系数。计算结果保留两位有效数字。五、验证结果1.线性结果将标准系列工作溶液分别注入液相色谱仪中,测定相应的峰面积,以标准系列工作溶液的质量浓度为横坐标,以峰面积为纵坐标,绘制标准曲线。同时做空白实验。6-苄基腺嘌呤[u]Y=74074.3*X-65.9497 R^2=0.9999959[/u][align=center][img=,595,372]http://ng1.17img.cn/bbsfiles/images/2018/07/201807242033263735_9846_2904018_3.png!w595x372.jpg[/img][/align]以上结果表明6-苄基腺嘌呤在0.04μg/mL~2.5μg/mL范围内,R[sup]^2[/sup]=0.9999959,6-苄基腺嘌呤浓度和峰面积呈线性关系,线性良好,符合要求。2.检出限结果将0.25μg/mL标准溶液逐级稀释至S/N=3±1,得出6-苄基腺嘌呤的方法检出限为0.0125mg/kg[color=#ff0000],[/color]此检出限结果小于国标GB/T 23381-2009的方法检出限0.02mg/kg,故此方法满足条件。六、方法精密度(重复性)对LBF180700282样品分别进行6次加标重复性的测定,测定结果如下:[table][tr][td][align=center]测定编号[/align][/td][td][align=center]1[/align][/td][td][align=center]2[/align][/td][td][align=center]3[/align][/td][td][align=center]4[/align][/td][td][align=center]5[/align][/td][td][align=center]6[/align][/td][/tr][tr][td][align=center]质量(g)[/align][/td][td][align=center]10.0031[/align][/td][td][align=center]10.0016[/align][/td][td][align=center]10.0025[/align][/td][td][align=center]10.0044[/align][/td][td][align=center]10.0027[/align][/td][td][align=center]10.0048[/align][/td][/tr][tr][td][align=center]浓度(μg/mL)[/align][/td][td][align=center]0.652[/align][/td][td][align=center]0.650[/align][/td][td][align=center]0.652[/align][/td][td][align=center]0.652[/align][/td][td][align=center]0.652[/align][/td][td][align=center]0.651[/align][/td][/tr][tr][td][align=center]含量(mg/kg) [/align][/td][td][align=center]0.33[/align][/td][td][align=center]0.32[/align][/td][td][align=center]0.33[/align][/td][td][align=center]0.33[/align][/td][td][align=center]0.33[/align][/td][td][align=center]0.33[/align][/td][/tr][tr][td][align=center]平均值(mg/kg)[/align][/td][td=6,1][align=center]0.33[/align][/td][/tr][tr][td][align=center]RSD%[/align][/td][td=6,1][align=center]1.24[/align][/td][/tr][/table]本方法的精密度为1.24%,符合GB/T 23381-2009中给出试样测试结果的精密度要求。因此,本次测定均符合要求。七、准确度验证(加标回收)对LBF180700282样品加标,取2.5μg/mL的标液0.09mL、0.35mL、0.64mL同样品同步处理后,结果见下表:[table][tr][td=2,1][align=center]测定编号[/align][/td][td=6,1][align=center]6-苄基腺嘌呤[/align][/td][/tr][tr][td][align=center]序号[/align][/td][td][align=center]m(g)[/align][/td][td][align=center]V(mL)[/align][/td][td][align=center]C(μg/mL)[/align][/td][td][align=center]6-苄基腺嘌呤含量(mg/kg)[/align][/td][td][align=center]平均值(mg/kg)[/align][/td][td][align=center]加标量(mg/kg)[/align][/td][td][align=center]回收率%[/align][/td][/tr][tr][td][align=center]1#[/align][/td][td][align=center]10.0236[/align][/td][td][align=center]5.0[/align][/td][td][align=center]N.D[/align][/td][td][align=center]N.D[/align][/td][td=1,2][align=center]N.D[/align][/td][td][align=center]/[/align][/td][td][align=center]/[/align][/td][/tr][tr][td][align=center]2#[/align][/td][td][align=center]10.0157[/align][/td][td][align=center]5.0[/align][/td][td][align=center]N.D[/align][/td][td][align=center]N.D[/align][/td][td][align=center]/[/align][/td][td][align=center]/[/align][/td][/tr][tr][td][align=center]加标1#[/align][/td][td][align=center]10.0087[/align][/td][td][align=center]5.0[/align][/td][td][align=center]0.042[/align][/td][td][align=center]0.021[/align][/td][td][align=center]0.021[/align][/td][td][align=center]0.022[/align][/td][td][align=center]95.5[/align][/td][/tr][tr][td][align=center]加标2#[/align][/td][td][align=center]10.0103[/align][/td][td][align=center]5.0[/align][/td][td][align=center]0.163[/align][/td][td][align=center]0.081[/align][/td][td][align=center]0.081[/align][/td][td][align=center]0.087[/align][/td][td][align=center]93.1[/align][/td][/tr][tr][td][align=center]加标3#[/align][/td][td][align=center]10.0189[/align][/td][td][align=center]5.0[/align][/td][td][align=center]0.307[/align][/td][td][align=center]0.15[/align][/td][td][align=center]0.15[/align][/td][td][align=center]0.16[/align][/td][td][align=center]93.8[/align][/td][/tr][/table] 由上表可以看出6-苄基腺嘌呤测定的加标回收范围在 60%-120% ,RSD值为1.31%符合规定要求。八、总结从检出限、线性范围、重复性、回收率测试结果可知,均符合方法要求,本实验方法符合GB/T 23381-2009的要求。[img=,595,372]http://ng1.17img.cn/bbsfiles/images/2018/07/201807242033072325_3650_2904018_3.png!w595x372.jpg[/img][img=,595,372]http://ng1.17img.cn/bbsfiles/images/2018/07/201807242033072325_3650_2904018_3.png!w595x372.jpg[/img][img=,595,372]http://ng1.17img.cn/bbsfiles/images/2018/07/201807242033072325_3650_2904018_3.png!w595x372.jpg[/img]
目前我们使用的自建库是西班牙一家公司的,但是我现在想把自建库的原料加上我们公司原料的编号,便于出报告方便调香师整理,但是目前我只是知道一个一个的编辑,是否可以批量编辑,是否可以把自建库以什么格式导出,然后编辑好后再导出?







 我要推广仪器
我要推广仪器
 下载APP
下载APP