我的PICO-TAG氨基酸法,前几天干燥氨基酸样品用的真空小瓶(WATERS干燥、水解氨基酸工作台专用的)、碎了[em63] ,做DNFB、DNCB效果都不理想,没有办法,只好请大家帮帮我,谁以前用过这个方法,淘汰的真空小瓶卖我一个。具体办法我们可以商量。我的QQ是78724851。电话13936003097。我很急,多谢了。[em48]
想咨询一下,0.1mol/L的PITC乙腈溶液取0.5ml,衍生化氨基酸溶液,氨基酸最好含多少mg啊?
哪位高手做过啤酒等含酒精饮料中氨基甲酸乙酯含量测定,请教啤酒中氨基甲酸乙酯含量范围,谢谢。
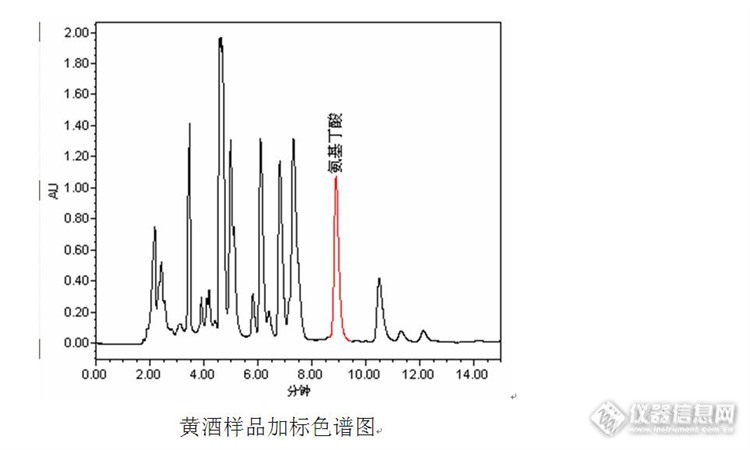
黄酒中γ-氨基丁酸含量测定的辛酸历程 近日实验室收到一批黄酒样品,该批黄酒是用发芽糙米为原料酿造而成,客户要求测定黄酒中的γ-氨基丁酸含量。由于之前实验室以丹磺酰氯为衍生试剂,建立了高效液相色谱法测定发芽糙米中γ-氨基丁酸含量的实验方法,并对实验方法的线性、精密度以及回收率进行了确认,均可以满足发芽糙米中γ-氨基丁酸含量测定要求,因此拿到黄酒样品后直接按照发芽糙米的前处理方法和色谱方法进行分析。链接如下:http://bbs.instrument.com.cn/shtml/20141226/5591256/。然而事与愿违,在测定的液相色谱图中压根就没有见到γ-氨基丁酸的色谱峰,反而在11.5min左右有个小的色谱峰,其峰高与发芽糙米中γ-氨基丁酸峰高有点相似,初步怀疑是保留时间发生了漂移,与发芽糙米样品色谱图对比后发现,在发芽糙米样品色谱图中该保留时间处也出现了一个相似的小峰,因此将该色谱峰是γ-氨基丁酸的可能性排除。http://ng1.17img.cn/bbsfiles/images/2014/12/201412311333_530568_1669358_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412311334_530570_1669358_3.jpg 原本该实验到此结束,准备将实验结果反馈给客户:黄酒中γ-氨基丁酸的检测结果为“未检出”。为了保证数据的准确性和可靠性,在黄酒样品中进行加标实验,结果在加标的色谱图中也未在相应的保留时间出峰,而且11.5min左右的色谱峰也没有增大,因此决定先将“未检出”的结果搁置,并对实验方法进行分析。 经过对样品前处理过程和色谱方法的分析,觉得可能造成加标样品中γ-氨基丁酸未检出的原因可能有:(1)保留时间漂移。由于流动相需要调节pH值,同时样品前处理过程中也涉及到酸、碱溶液的使用,怀疑是流动相或者样品pH的改变导致保留时间的漂移,从而未在原有的保留时间出现应有的色谱峰。然而重新配制流动相和前处理样品,加标样品测定结果依然是“未检出”,对比加标和不加标样品的色谱图,两者几乎一样,也没有峰面积或峰高变化明显的色谱峰;(2)衍生试剂失效。丹磺酰氯对光和湿敏感,不稳定,放置时间久了会生产二氯亚砜并继续分解成其他物质,影响其在有机溶剂中的溶解度,也会影响结果。可是为了排除衍生试剂的问题,重新打开一瓶刚购置不久的丹磺酰氯试剂,并重新试验,结果仍然不理想;(3)衍生条件控制不当。之前用相似的方法测定牛磺酸含量以及测定发芽糙米中γ-氨基丁酸含量时曾出现过衍生过程条件控制不当造成衍生不完全或者不能衍生的情况,可是与黄酒样品同一批处理的γ-氨基丁酸标准溶液和发芽糙米样品均能衍生成功,并正常出峰,为何唯独黄酒样品不出峰呢?在百思不得其解之际,看到同事在滴定黄酒中总酸,忽然间若有所悟:黄酒中的γ-氨基丁酸需要在碱性条件下才能与丹磺酰氯发生衍生反应,而黄酒是酸性介质,pH值一般在3~5之间,同时黄酒为酿造产物,对酸碱性具有一定的缓冲能力。http://ng1.17img.cn/bbsfiles/images/2014/12/201412311336_530572_1669358_3.jpg 通过比较发现:黄酒为酸性样品,缓冲能力较强,按照发芽糙米样品前处理方法直接加入0.5mL 碳酸钠(pH9.8)可能不能达到合适的衍生反应条件,最终导致黄酒样品中γ-氨基丁酸“未检出”。 找到问题后调整实验方案,先将黄酒样品调整至中性,然后再按照发芽糙米样品方法进行前处理。调整实验方案后,黄酒样品中γ-氨基丁酸测定的色谱图如下图。从色谱图中可以发现,经过实验方案的调整黄酒样品中检出了γ-氨基丁酸的存在。http://ng1.17img.cn/bbsfiles/images/2014/12/201412311337_530573_166
请问各位专家,我在做白酒氨基甲酸乙酯时,用的安普的白酒氨基甲酸乙酯固相萃取小柱子,出来的图谱在目标物保留时间附近有峰,但是离子比例不对。不知道该如何定性。中间试过加标,如果浓度大了,会把原峰覆盖,离子比例就匹配了,如果浓度不高,依旧是离子比例不匹配。
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=155679]小麦籽粒氨基酸碳氮稳定同位素的测定与分析[/url]………………………………………………………………………………[color=#00008B]【目的】利用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-燃烧-同位素比值质谱仪(gas chromatography-combustion-isotope ratio masss pectrometry,GC-C-IRMS)测定小麦籽粒氨基酸碳氮稳定同位素组成。【方法】以小麦临汾50744为材料,水解得到其籽粒蛋白质氨基酸,将氨基酸标准样品以及小麦籽粒氨基酸衍生化为N-新戊酰基,O-异丙醇(N-pivaloyl-isopropyl,NPP)氨基酸酯,利用GC-C-IRMS测定其碳氮稳定同位素组成。【结果】氨基酸标准样品的碳氮同位素组成分析表明,NPP氨基酸酯的平均重现性δ^13C为0.47‰,δ^15N为0.28‰,并没有产生大的同位素分馏,因此δ^13C和δ^15N都能得到满意的测定结果。运用GC-C-IRMS测定了小麦临汾50744籽粒蛋白质氨基酸的稳定碳氮同位素的自然丰度,其中δ^13C的变化范围在-28.7‰到-34.7‰,δ^15N的变化范围为-6.2‰到9.5‰。采用系统聚类分析进行分类,根据δ^13C可以将氨基酸分为两类 根据δ^15N可以将氨基酸分为三类。【结论】运用GC-C-IRMS结合NPP氨基酸酯衍生物可以测定小麦籽粒氨基酸的稳定碳氮同位素,这对于揭示氨基酸代谢途径的差异以及逆境胁迫下氨基酸的合成差异具有重要的意义。[/color]
关于啤酒中的氨基甲酸乙酯含量检测谁有标准和检测方法?
[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]测氨基丁酸的文献
最近在做PITC柱前衍生测定18种氨基酸。但是老是发现图谱第三分钟出现一个非常高的峰,并且随着样品稀释浓度降低而降低。很郁闷。各位朋友,如果做过此类实验的话,请给予指教。不甚感激!
滴定保护氨基酸?
请教滴定保护氨基酸的方法?
求助各位高手,在什么情况下需要对皮革测4-氨基偶氮苯?
最近准备做氨基甲酸乙酯,用之前的标准品,用之前的方法做不出峰来,用1ppm的全扫也不出峰之前是同事做的,不知道不出峰会是什么原因?是标准品不稳定分解掉了,还是现在仪器状态不如以前了?有没有做过氨基甲酸乙酯的朋友,请多多指教!谢谢
碱液滴定氨基酸时,加入甲醛是什么原理
有人做过2氨基丁酸的衍生吗?求助
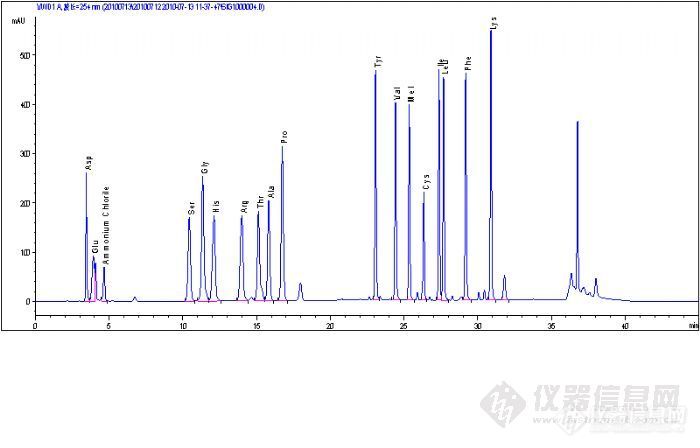
大家好: 最近在做PITC氨基酸柱前衍生,氨基酸标准图谱已经完全跑出来了,但是样品的图谱不是很好看,最后八个峰老是跑不出来,紧急求助大家!我的QQ306157758,如果可以联系我,麻烦你们了!我已经把图谱上传。我怀疑是样品没有完全水解,我用的螺旋盖的离心管去水解。http://ng1.17img.cn/bbsfiles/images/2010/09/201009020958_240923_1689001_3.jpghttp://ng1.17img.cn/bbsfiles/images/2010/09/201009020959_240924_1689001_3.jpg
有哪位高人测过γ-氨基丁酸,我查到的标准是QB/T4587-2013,具体操作中,有哪位高人做过,帮忙指导一下,有哪些注意的,谢谢
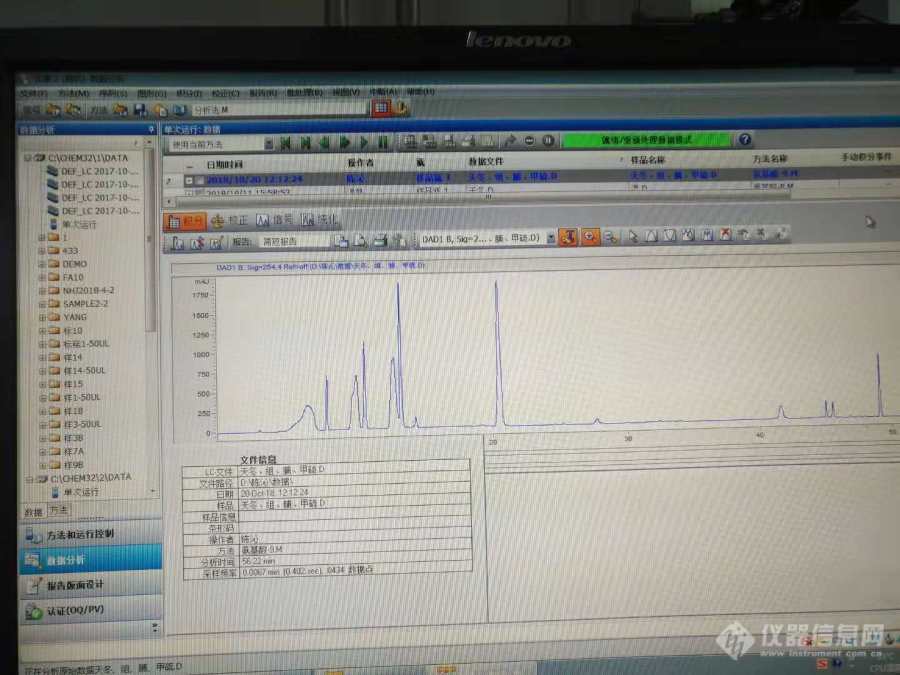
我是参考一篇PITC柱前衍生法测黄酒中氨基酸含量的文献,方法、色谱柱都和文献一样,但是为什么我跑出来的色谱峰有很多都是一种氨基酸对应两个色谱峰。我每次衍生剂都是现配现用的,我换过一根色谱柱,结果还是一样,我不知道问题出在哪里。请大家帮忙分析分析原因,十分感谢。如图1是我跑的四种氨基酸的标品,前面两种氨基酸都是分别对应两个色谱峰。后面两个氨基酸分别是单独的一个峰。图2是我跑的17种氨基酸混合标品.后面两张图是我参考的文献里面的具体洗脱方法.请各位大神给予指点.[img=,554,296]https://ng1.17img.cn/bbsfiles/images/2018/10/201810240941270206_3458_3363167_3.png!w554x296.jpg[/img][img=17种氨基酸混合标品,554,296]https://ng1.17img.cn/bbsfiles/images/2018/10/201810240936371461_9895_3363167_3.png!w554x296.jpg[/img][img=,428,311]https://ng1.17img.cn/bbsfiles/images/2018/10/201810240940035288_3112_3363167_3.jpg!w428x311.jpg[/img][img=参考的文献具体洗脱程序,435,294]https://ng1.17img.cn/bbsfiles/images/2018/10/201810240936544035_7123_3363167_3.jpg!w435x294.jpg[/img]
急!最近在做18氨基酸查了一些资料,最后选择了异硫氰酸苯酯(PITC)衍生测18氨基酸但遇到不少问题,希望得到大家帮助!主要体现在丝氨酸、甘氨酸、组氨酸、精氨酸。。。。到脯氨酸不能很好的分离开,且峰形不好看理论板数不高,后面9个氨基酸又能很好的分离峰形好不知道是那里出了问题?流动相还是衍生过程?不知道你们能否提供一份成熟的方法,我好做下对比找到问题所在在此感谢!可发邮箱:liujian132008@yahoo.com.cn也可以提供意见!大家一起讨论讨论。
由于实验的需要,需要液体样中引入氨,后来通过旋蒸尽量的除去了氨气,但还是有部分残留使样品的PH保持在8.0左右,请问:这会不会影响氨基态氮的滴定 ?我个人认为是不影响的,想听听大家的看法!
[color=#444444]本人最近做氨基酸的PITC柱前衍生试验,其中一个标品谷氨酸出峰在3-4min出来,但是峰分叉,感觉不是一个物质,请问下有相关经验的老师指教一下,本人万分感激![/color][color=#444444]色谱条件为:[/color][color=#444444]柱子:Agilent SB-C18 150mm*4.6mm, 5um[/color][color=#444444]流动相A:0.1mol/L NaAC (pH=6.5)-乙腈(97:3)[/color][color=#444444]流动相B:乙腈:水=4:1[/color][color=#444444]进样量10uL[/color][color=#444444]柱温40℃[/color][color=#444444]结果谷氨酸峰形分叉,前肩峰拖得厉害,非常难看,后面其他氨基酸的峰形正常,并且能够分离。[/color]
[color=#444444]本人最近做氨基酸的PITC柱前衍生试验,其中一个标品谷氨酸出峰在3-4min出来,但是峰分叉,感觉不是一个物质,请问下有相关经验的老师指教一下,本人万分感激![/color][color=#444444]色谱条件为:[/color][color=#444444]柱子:Agilent SB-C18 150mm*4.6mm, 5um[/color][color=#444444]流动相A:0.1mol/L NaAC (pH=6.5)-乙腈(97:3)[/color][color=#444444]流动相B:乙腈:水=4:1[/color][color=#444444]进样量10uL[/color][color=#444444]柱温40℃[/color][color=#444444]结果谷氨酸峰形分叉,前肩峰拖得厉害,非常难看,后面其他氨基酸的峰形正常,并且能够分离。[/color]
有没有试用氨基柱的同仁啊?最近我买了跟国产的氨基柱做糖,流动相为乙腈:水=65:35,各个组分保留时间老是不稳定,而且呈有规律的变化。变化规律为每一次进样都比前一次进样晚出峰0.5min左右。各位老大,谁能帮忙解释一下啊???????
最近我们在滴定对氨基苯酚时问到了个头疼的问题,根据国标称取试样约0.25-0.30g(精确至0.0001g),置于250mL烧杯中,加入150mL水、10mL盐酸、10ml溴化钾,使样品完全溶解。冷却至10℃~15℃,然后将滴定管尖端插入溶液中,在不断搅拌下,用亚硝酸钠标准滴定溶液进行滴定,滴定近终点时把滴定管尖端提离液面,继续滴定直至使淀粉-碘化钾试纸呈现微蓝色润圈,并保持3min不变即为终点,在相同条件下做空白试验。在我滴第一滴时,整个溶液就显蓝色,严重时显蓝紫色。再滴定时蓝色会逐渐消失. 我刚开始以为是碘离子和淀粉。我重新称取样品溶解,直接加入碘溶液,溶液不变蓝,证明样品里没有淀粉,如果样品里没有淀粉,就证明变蓝的不是碘离子。所以我想请教大家什么物质在酸性条件下,与亚硝酸钠溶液发生反应会变蓝色,继续滴定蓝色会消失?
请教一下各位:用氨基酸分析仪能检测皮革水解蛋白吗?
本人用PITC衍生法测17种氨基酸,跑单一标品时胱氨酸出现好几个峰,且与混标对应不上,想问大家是否有做出来的?需要预处理吗?说明一下,本人用的是岛津的c18柱子(25cm),液相也是岛津的,氨基酸混标为安捷伦的。请求大家帮忙。
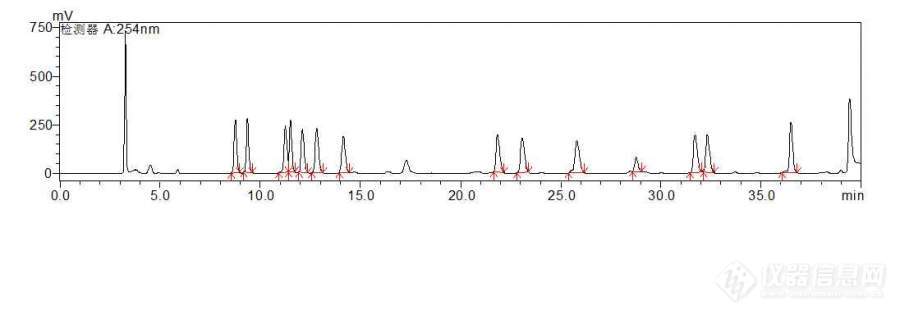
[color=#444444]如题。用的是安捷伦C18柱子,岛津15C系统。跑了几个不同浓度的氨基酸标品后,5min以前应该出现两个氨基酸峰的,现在发现是出来[/color][color=#444444]一个大峰,怀疑是两个氨基酸没有分离开。会是什么原因呢?以前做过是分离开了,但是现在却没分开来,我用的标品是新买的。。。急啊!是PITC放太久了吗?[/color][color=#444444][color=#444444]这是跑的一个标品图谱。30-35min的一个峰也没有跑出来。怎么回事呢?[/color][img=,690,241]https://ng1.17img.cn/bbsfiles/images/2019/06/201906281553375379_8519_1847709_3.jpg!w690x241.jpg[/img][/color]
求救!!要用GC做空气中丁氨基乙醇,请问用什么解吸液可以把碳管解析出来啊~~~标液用什么溶剂配制啊!多谢!
最近在用异硫氰酸苯酯(PITC)检测氨基酸的时候出现了一个怪问题:17个氨基酸只有前面9种氨基酸可以出峰,剩下的氨基酸都不能出峰。液相条件什么都是一样的,换了新柱子问题仍然存在。一开始怀疑由于现在天气凉了衍生不充分,就放在25℃水浴中衍生2小时,问题仍然存在。让我很是苦恼。调整梯度程序、温度都不管用。液相也很正常。下面是我的液相条件:请各位高手帮忙啊!5色谱条件色谱柱: Sepax AA专用柱,4.6*250mm,由苏州赛分科技有限公司提供。检测波长:254nm。柱温:40℃。流动相A: 醋酸钠溶液:乙腈=93:7 (V/V)。流动相B: 乙腈:水=4:1(V/V)。进样量:5ul流速:1mL/min(注:分析结束后使用20%乙腈水溶液清洗,再用100%乙腈清洗后储存) 表1 梯度洗脱程序 时间/min 流动相A/% 流动相B/% 0 100 0 2 100 0 14 93 7 29 70 30 32 50 50 33 0 100 39 0 100 39.1 100 0 45 100 0 6 氨基酸标准溶液和样品溶液的衍生(设2个重复)6.1 17AA衍生准确量取氨基酸标准溶液400μl,置5.0ml塑料离心管中,加入三乙胺乙腈溶液(2.10)200μl,异硫氰酸苯酯乙腈溶液(2.11)200μl,混匀,室温放置1小时,然后加入正己烷800μl,振摇后放置10分钟,取下层溶液(PTC-AA),用一次性注射器吸取上清液用0.45μm针式过滤器过滤。准确量取样品溶液400μl,置5.0ml塑料离心管中,加入三乙胺乙腈溶液(2.10)200μl,异硫氰酸苯酯乙腈溶液(2.11)200μl,混匀,室温放置1小时,然后加入正己烷800μl,振摇后放置10分钟,取下层溶液(PTC-AA),用一次性注

氨基酸态氮在食品理化检验中为较常见的检验项目,酱油,黄酒,调味酱料、豆腐乳,果汁等样品中,均涉及该项目的检测。传统做法在于,全用手工移液滴定。首先先用滴定管滴到8.2,再用移液管移取10mL的甲醛溶液加入溶液中,再用滴定管滴到9.2。过程较为麻烦。本科室目前采用2种方法来解放手工操作。1. 样品量大的时候, 利用带转盘(自动进样)的电位滴定仪,设定程序后,全程自动滴定。这种虽然全程无人值守,但时间耗费较多。2. 样品量较小的时候, 用下面这种装置组合。由于人为判断和操作,所以速度较快。本文着重介绍本科室样品量较小的时候使用的装置组合。一种高效滴定食品中氨基酸态氮的装置组合,其特征在于包含以下装置:1. pH计及其水系电极;2. 转子及其磁力搅拌器;3.数字滴定器;4.分液器;具体如下图所示;http://ng1.17img.cn/bbsfiles/images/2016/08/201608121403_604658_2240076_3.jpg图中,分液器下面装的是分析纯的甲醇溶液。右边为数字滴定器,下面绿色塑料罐装的是氢氧化钠标准滴定液。浓度可以为0.1或0.05mol/L.根据实际需要填装。蓝色的为磁力搅拌器。近距离看http://ng1.17img.cn/bbsfiles/images/2016/08/201608121403_604659_2240076_3.jpg实际操作的时候。先装好装置,把电极(已校准好)深入溶液中,将分液器和滴定器的管头深入杯中并悬空,处于待滴状态。打开磁力搅拌,用数字滴定器先滴到8.2后,直接拉升按下加入10mL的甲醛溶液后,再滴到pH9.2后。该装置组合的优点在于, 固定各装置的位置以后,整个实验过程方便并且流畅。另外,做批量样品的时候,换下一个样品的速度也很快。。分液器不需要手工移液,减少了实验过程中人员同甲醛试剂的接触;数字滴定器不需要手工加液,只需要按下数字清零键即可继续滴定;减少了加液、调零、读数的时间,并且数字显示,降低人为目视误差。(该数字滴定器经过计量,相当于A级玻璃滴定管的准确度)







 我要推广仪器
我要推广仪器
 下载APP
下载APP