最近刚开始做[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url],对于很对东西都是边学边做,对衍生化不太熟悉,我用的是BSTFA-TMCS加吡啶70℃下水浴30min,进样1微升,跑了混合的,出了四个峰(不知道17分钟之后那个小的算不算,响应值低于25)我就以为正好,结果单个进样的时候对不上,有没有人做过这个呀,或者有没有大佬说一下可能的原因呢,下面依次是混合,月桂酸,月桂酸单甘脂,三月桂酸甘油酯,三丁酸甘油酯。[img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206171033356375_1876_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206171033355547_6193_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206171033356248_8395_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206171033355761_4184_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206171033356826_8013_5654520_3.png[/img]
二月桂酸二丁基锡是否禁用?
二月桂酸二丁基锡是否禁用?
二丁基二月桂酸锡和二丁基二醋酸锡等有机锡类能否用某种特定的色谱柱检测?
国标乳品中碘的测定中,在硫酸条件下碘与丁醇反应生成丁醇与碘的衍生物,到底此衍生为何物?
最近刚开始[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url],对衍生化不太了解,我用的是BSTFA+TMCS进行衍生化的,混合的出了四个峰(最后那个17分钟之后的不知道算不算峰,响应值<25),我以为正好,结果跑单个的对不上了,下面的图依次是混合,月桂酸,月桂酸单甘脂,三月桂酸甘油酯,三丁酸甘油酯。有没有做过的呀[img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206170905473939_5594_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206170905475042_7945_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206170905475286_2664_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206170905475296_4906_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206170905475101_6177_5654520_3.png[/img]
最近在做关于壬二酸、癸二酸和十一碳二元酸的点板分离,目前使用了一些展开剂,基本没有移动,不知哪位老师做过类似实验,指导一下,谢谢!
关于乙醇、乙酸乙酯、甲基异丁酮、甲苯、丁酸乙酯、异戊酸乙酯、乙酸-2-甲基-1-丁醇酯、己酸乙酯、异戊酸异戊酯、异丁酸异戊酯、己酸烯丙酯、紫罗兰酮、肉桂酸异丙酯13种物质的气相方法,这13种物质能否一针全部出来,用什么柱子合适,FID对它们是不是都有响应?希望各路高手路过能留下点意见和相关资料,不胜感激!
摘要:建立胡黄连中香草酸和桂皮酸的含量测定方法。方法用双波长扫描法测定胡黄连中香草酸和桂皮酸的含量。结果香草酸。桂皮酸斑点峰面积3Il内稳定,香草酸回收率为103.86%,RSD=1.33%,桂皮酸回收率为103.16%,RSD=1.28%。结论该方法稳定,可行。具有实用性。 关键词:胡黄连 薄层扫描法 香草酸 桂皮酸 胡黄连具有保肝利胆、抗炎、抗真菌等药理作用。胡黄连含胡黄连素、胡黄连苷(I II III)、D-甘露醇、香草酸、肉桂酸、胡黄连醇成分。香草酸和桂皮酸是其中的两种抗菌成分。我们对胡黄连中香草酸、桂皮酸含量建立了薄层扫描法,以达到控制胡黄连的质量,从而为临床疗效提供保证。 1 仪器与试剂 药材:胡黄连,太原市药材公司;仪器:日本岛津CS--9301PC薄层扫描仪;手提式荧光灯(上海固村电光仪器厂);对照品:香草酸对照品(中国药品生物制品检定所);桂皮酸对照品溶液(省药检所提供e=0.604mg/50ml);硅胶GF254(青岛海洋化工厂)所用试剂均为分析纯。 2 实验条件 2.l 薄层层析条件:分别以石油醚-氯仿-丙酮-冰醋酸(10:4.4:10.1);正己烷-乙醚-冰醋酸(5:5:0.1);正己烷-氯仿-乙醚-冰醋酸(5:3:2:0.1)以及氯仿:甲醇(2:1)展开,多次比较发现正己烷。氯仿-乙醚-冰醋酸(5:3:2:0.4)分离效果好。 2.2 测定波长及主要扫描参数,分别对香草酸,桂皮酸对照品斑点在200nm-370nm扫描,在290nm处有最大吸收,350nm处无吸收,固定350nm为参比波长,290nm为测定波长。
硅氟酸钾法测定硅为什么不稳定,每次数据不一样?
硅酸甲酯加到香精中会有沉淀析出。供应商解释说析出了桂酸。取单独的硅酸甲酯,一份加丙酮,一份加乙醇,沉淀都不溶。是什么原因呢?有遇到同样问题的亲没?
用邻苯二甲酸氢钾标定的氢氧化钠,算其对于硅氟酸的滴定度,应该怎样计算,谢谢!
目前实验室配备的气相色谱只有FID,现在想检测碘丙炔醇丁基氨甲酸脂(CAS:55406-53-6),找到一份文件上说要用ECD检测,请问各位大虾,有没有人做过这个实验,用FID可以检测吗?谢谢了!
我使用的是日本AND HR200型電子天平,精確到0.1毫克,今天突然發現歸零後最後一位數字總是不穩定,一直跳動,各位知道是怎麼回事嗎???
如题,硅钨酸重量法测定维生素B1的含量,请问各位老师,其中的原理。整个过程哪些因素影响结果?维生素B1在酸性溶液中,与硅钨酸作用生成沉淀……维生素B1与硅钨酸摩尔比是多少?反应方程式是怎样的?酸性溶液中,那么酸性溶液如何控制?pH在什么范围最利于反应进行?反应时间是否需要控制?多长时间?
各位老师 钢铁及合金钢中硅的状态还分为“酸溶硅”和“全硅”啊二者含量差的多么 咱么做试验所带的标样中硅的含量一般指的是酸溶硅还是全硅?全硅是酸溶硅和酸不溶硅的合么?紧急啊....
硅钼杂多酸有α和β两种形态。α型在较低酸度下(pH2.3~3.7)的热溶液中生成,很稳定,其最大吸收波长为314nm,ε=17000;还原后为蓝绿色,最大吸收波长为635和750nm,ε=18500。β型在较高酸度(pH1~2)的溶液中生成,最大吸收波长为312nm,ε=21000;还原后为蓝色,最大吸收波长为810nm,ε=20000。β型不稳定,容易转变成α型,转变速度随温度升高、酸度降低及溶液中离子强度增大而加快。由于硅钼杂多酸还原产物ε值不同,吸光度随分析条件而异。当用硅钼黄光度法时适宜采用α型,为此在pH3.0~3.7酸度条件下,而且沸水浴5~10min,以保证β型转变成α型。硅钼黄光度法实际使用范例为铝合金中硅的测定。当用硅钼蓝光度法时适宜采用β型,因为它在较高酸度下生成,铁不会形成沉淀,而且它比α型容易还原。为了避免β型转变成α型,必须控制β型的络合时间及温度,在pH1.5时,沸水浴30s即可,如果时间太长,将由于转变成α型而使吸光度降低。硅钼蓝光度法实际使用范例很多,如钢铁中硅的测定。
分析月桂酸,月桂酸单甘脂,三月桂酸甘油酯用什么色谱柱比较好啊,目前用rtx1好像分不开,我用的BSTFA+TMCS衍生化后分析的
用硫代硫酸钠滴定碘伏消毒剂,当溶液变成浅黄时加入淀粉溶液,不变色,没有蓝色出现。以前滴定碘类消毒剂偶尔也会遇见这种情况。请教各位大侠这是怎么回事?我滴定的这个碘伏溶液是聚醇醚碘,由脂肪醇聚聚氧乙烯醚、碘、碘化钾、醋酸配一起络合而成。我在想会不会是配制的碘伏溶液的某些因素影响到淀粉变蓝?各位大侠有知道的吗?跪求答案。
本人实验中要分开γ-丁内酯、1,4-丁二醇和丁二酸二甲酯,但γ-丁内酯和丁二酸二甲酯可以分,加入少量的1,4-丁二醇也可以分,但一旦量大了1,4-丁二醇就显示两个峰,并且如果继续加1,4-丁二醇则又会变为一个峰,试问高手这是怎么回事呀?是不是我条件有问题呀,我用的是程序升温。非极性柱子
哪位老师有月桂酸的标准。

图文再现经典方法中的经典“高氯酸脱水重量法测定硅铁中的硅” 重量法测定硅的关键,在于脱水是否完全。用盐酸脱水效果较差,需进行二次脱水;硫酸脱水比盐酸要好一些,但冒烟时产生飞溅;高氯酸脱水所得二氧化硅较纯净,在日常分析中,对于低含量的硅,一次脱水脱水即可。对于高含量的硅,则需要二次脱水,而且数据的稳定性好,绝对是经典方法中的经典方法!!下面就结合图给大家再现一下经典方法。1 方法原理用过氧化钠-碳酸钠混合熔剂熔融试料,盐酸酸化,高氯酸二次冒烟,使胶体的硅酸脱水,形成不溶性的硅酸。沉淀于1000~1100℃灼烧至恒量。加硫酸、氢氟酸处理,使硅呈四氟化硅逸出,再灼烧至恒量。由加氢氟酸处理前后的质量差计算硅的质量分数。2 试剂2.1 混合熔剂,过氧化钠+碳酸钠=2+12.2 盐酸,r约1.19g/mL2.3 高氯酸,r约1.67g/mL2.4 氢氟酸,r约1.15g/mL2.5 盐酸,1+1、5+952.6 硫酸,1+12.7 硝酸银,10g/L3 操作步骤3.1 称样首先天平要注意归零。称取0.20000g粒度小于0.125mm的试样,精确至0.0001g。随同试料做空白试验。http://ng1.17img.cn/bbsfiles/images/2012/12/201212051927_409653_1607403_3.jpg 天平万分之一的,绝对称量有保证!3.2 试料处理将试料置于盛有6g混合熔剂的镍坩埚中,混匀,再盖2g混合熔剂。先低温烘焙至焦黄色,再于850°C的高温炉中或喷灯上熔融至透明。取出稍冷,用水吹洗净坩埚外壁。将坩埚放入[si
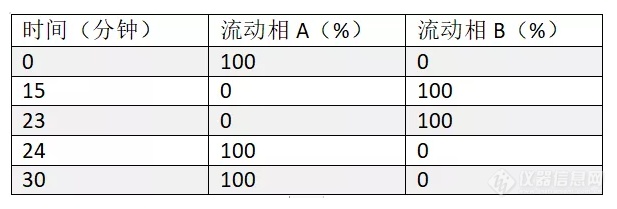
[align=center][b]盐酸雷尼替丁系统适用性试验-2015中国药典[/b][/align]色谱条件色谱柱:Kromasil 100-5-C18, 4.6*250mm货号:M05CLA25流动相A:酸盐缓冲液(取磷酸6.8ML置1900ML水中,加入50%氢氧化钠溶液8.6ML,加水至2000ML,用磷酸或50%氢氧化钠溶液调节pH 值至7.1±0.05):乙腈=98:2流动相B:磷酸盐缓冲液-乙腈(78 : 22)梯度程序:[img=,617,209]https://ng1.17img.cn/bbsfiles/images/2018/11/201811131416404930_2830_2428063_3.png!w617x209.jpg[/img]流速:1.5ML/min柱温:35℃波长:230nm进样量:10μL[img=,534,227]https://ng1.17img.cn/bbsfiles/images/2018/11/201811131416589880_2883_2428063_3.png!w534x227.jpg[/img]结论:1. 出峰顺序为杂质I,雷尼替丁2. 杂质I的相对保留时间约为0.853. 雷尼替丁峰和杂质I峰的分离度大于4.0以上指标都符合药典要求。本应用来源于Kromasil微信公众号
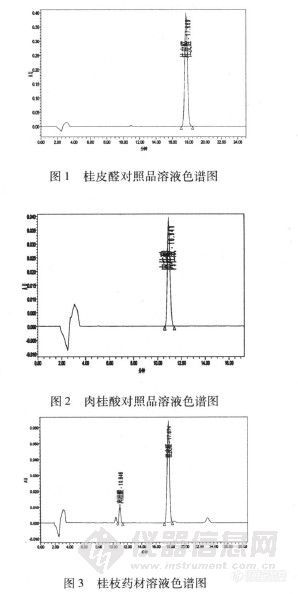
【作者】 王连芝; 蒋维谦;【机构】 黑龙江中医药大学中医药研究院;【摘要】 目的:建立HPLC法测定桂枝中桂皮醛和肉桂酸含量。方法:采用Diamonsil C18(250mm×4.6mm,5μm)色谱柱,以乙腈-0.1%磷酸溶液(38:62)为流动相,流速为1.0ml.min-1,检测波长为276nm和289nm双波长扫描。结果:样品中桂皮醛的平均回收率为99.48%,RSD为1.21%;肉桂酸的平均回收率为98.76%,RSD为1.29%;桂皮醛在0.01~0.03之间峰面积与浓度线性关系良好(r=0.9998);肉桂酸在0.002~0.01μg之间峰面积与浓度线性关系良好(r=0.9997)。结论:该实验方法简便,重现性好,回收率高,可作为同时测定桂枝中桂皮醛和肉桂酸含量的方法。 更多还原【关键词】 桂皮醛; 肉桂酸; 高效液相色谱法; 桂枝; 【基金】 黑龙江中医药大学科研基金项目(200745)http://ng1.17img.cn/bbsfiles/images/2012/08/201208071034_382135_2352694_3.jpg
求助月桂酸钾检测标准,谢谢!
硫酸沙丁胺醇 欧洲药典8.8
摘要:目的 建立一种快速测定参枝苓口服液中肉桂酸含量的方法。 方法 首先采用高效液相色谱法测定参枝苓口服液中肉桂酸含量,做为一级数据,并同时采集样品的近红外光谱;采用K-S方法对样品集进行划分考察不同的光谱预处理方法,以Rc、Rv、RMSECV、RMSEP值为模型评价指标。 结果 最佳模型Rc、Rv、RMSECV、RMSEP值分别为0.8862、0.8877、0.00282、0.00301。线性较好,光谱与样品的肉桂酸含量有较好的相关性。结论 建立的参枝苓口服液中肉桂酸含量模型满足生产需求,是一种快速无损、经济的检测方法。关键词:近红外光谱分析技术 参枝苓口服液 肉桂酸 参枝苓口服液是我国首个批准用于老年痴呆的中药复方药物,适用于轻中度阿尔茨海默病所引起的心气虚证,表现症状为心慌心悸、气虚少语、神情冷漠、头晕乏力、失眠健忘、舌淡脉虚等症。组方包括党参、桂枝、茯苓和白芍等10味中药,其中桂枝是主要药材之一,桂枝中主要含有肉桂酸、桂皮醛等成分在药效中起到关键作用。因此,可以通过检测复方中肉桂酸的含量来检验和控制参枝苓口服液的药品质量。目前使用的含量检测方法为HPLC法,该方法比较复杂,不利于实现在线控制。近红外光谱分析技术(Near Infrared Spectroscopy,NIRS)是当前最重要的一种PAT技术,它是指应用波长范围在780-2526nm波长范围内的电磁波的一种快速绿色无损光谱学分析方法并且已经开始广泛应用于农副产品检测、食品检测、药物检测、石油化工产品检测、烟草检测等领域。本实验采用NIRS方法对复方中肉桂酸含量进行测定。1仪器与材料1.1试剂乙腈、甲醇为色谱纯(Merck, Darmstadt, Germany),磷酸为色谱纯(Tedia公司)。肉桂酸对照品购自中国药品生物制品检定所(北京),其余试剂均为分析纯。1.2样品收集四批(生产批号分别为14311、14312、14313、14314)共85个样品,所有样品均有潍坊沃华医药有限公司提供。1.3仪器Agilent 1200高效液相色谱仪,四元泵、DAD检测器、ALS自动进样器、柱温箱Chem-Station for LC 3工作站;柱子型号:Aglient Eclipse Plus C18柱(4.6×250mm,4.6um);Millipore超纯水器(美国Millipore公司);AntarisⅡ傅里叶变换近红外光谱仪(美国Thermo Fisher公司),配有透射模块采样系统和Result操作软件;其他玻璃仪器。光谱处理及模型建立采用Matlab 软件和TQ 软件。2方法2.1近红外光谱的采集采集光谱所用的近红外光谱仪器为美国Thermo Fisher公司生产的AntarisⅡ傅里叶变换近红外光谱仪;将适量样品置于4mm的玻璃样品管中进行光谱采集,光谱采集范围为4000 -10000 cm-1;扫描次数为32;分辨率为8 cm-1;增益值为4x,常温环境采集光谱。每次采集样品前采集背景光谱来消除背景的影响。每个样品的测量时间小于1 min。2.2 HPLC测定芍药苷含量本实验采用HPLC方法作为参考方法来测量样品肉桂酸含量的一级数据。以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.1%磷酸溶液(25:75)为流动相,待肉桂酸色谱峰出峰后,再用乙腈为流动相洗脱10分钟;检测波长为278nm。理论板数按肉桂酸峰计算应不低于12000。取肉桂酸对照品适量,精密称定,加50%甲醇溶液制成每47.1μg/ml的标准品溶液,即得对照品溶液。精密量取本品10ml,置20ml量瓶中,加甲醇稀释至刻度,摇匀,离心,取上清液,即得供试品溶液。分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,按上述色谱条件,1ml/min流速下等度洗脱50min,记录对应峰面积,外标一点法计算含量。2.3近红外光谱技术分析采用PLS方法将样品液相一级数据与近红外光谱(二级数据)相关联,通过光谱预处理方法的选择、光谱建模区间的选择以及其他建模参数的优化,建立稳健的定量分析模型,并以模型的RMSEC、RMSEP、Rc和Rv值作为模型结果的评价指标。2.3.1校正集和验证集的划分所有的样品划分为校正集和验证集。样品集划分的方法有随机样品划分法(RS),KS法以及SPXY法。在本实验中采用KS法将样品划分为校正集和验证集。KS方法是基于光谱变量的选择方法。根据约4:1的比列将样品划分为68个校正集和17个验证集。2.3.2光谱建模区间的选择本实验分别采用全光谱(4000-10000cm-1)区间以及TQ软件所建议优化的区间(8327-5457cm-1)建立样品的PLS,以Rc、Rv、RMSECV、RMSEP值为参数对模型进行比较,选出最佳的建模光谱区间。2.3.3光谱预处理方法的选择近红外光谱中包含复杂的样品化学信息,在分析过程中,近红外光谱受多种因素的干扰。在建立校正模型之前需要采用化学计量学的方法对光谱作适当的预处理以减少或消除这些影响,改善模型的性能。本实验分别采用SG15、MSC、SNV以及SG15+1d对原始光谱进行预处理并建立PLS数学模型。以Rc、Rv、RMSECV、RMSEP值为参数对模型进行比较,选出最佳的预处理方法。2.3.4模型的建立与模型的评价选用最佳的预处理方法和优化的光谱区间建立PLS模型,用验证集样品对最终模型进行外部验证,并以Rc、Rv、RMSECV、RMSEP值作为模型性能的评价指标。3结果3.1参枝苓口服液肉桂酸含量测定HPLC数据分析表1 参枝苓口服液肉桂酸含量测定统计表 [col
请问有朋友遇到过SPME顶空瓶(隔垫)带来硅氧烷(流失)的吗?
[color=#444444]有没有人检测过买来的月桂酸的质量?采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法?里面含有哪些有关物质?[/color]

作者:李昂; 胡翮; 郭丙炎;(湖南省湘潭市药品检验所;)摘要:目的用反相高效液相色谱(RP-HPLC)法测定盐酸丁卡因的含量。方法色谱柱为Diamonsil C18柱(150mm×4.6mm,5μm),流动相为水-甲醇-乙腈(45∶45∶10,v/v,含0.02%庚烷磺酸钠,0.34%磷酸二氢钾,三乙胺调pH至7.0),流速为1.0mL/min,检测波长为314nm,柱温为室温。结果盐酸丁卡因质量浓度在5.034~161.1μg/mL范围内与峰面积线性关系良好,回归方程为Y=5.6572×105X-9.2039×105(r=0.9999),平均回收率为100.76%,RSD为0.41%(n=6)。结论RP-HPLC法简便、可靠、准确,可作为盐酸丁卡因注射液的含量测定方法。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208271103_386360_1606903_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP