大家知道,长叶蒎烯LONGIPINENE和长叶烯 Longifolene 在哪些天然精油(食用香精)里面含量比较高。
昨天看到几个未知物求定性的帖子,经过朱老师的帮忙 确认为 isolongifolenlactone 昨天用自动进样 搞了几个 异长叶烷酮 证实此物质确实是 来自异长叶烷酮 但是有个疑问···············大多数的异长叶烷酮是:2个主峰(质谱能定性的) 先小后大但是我发现有一个异长叶烷酮的峰形是 先大后小 而且,这个“异长叶烷酮”的气味也比其他的异长叶烷酮气味要好的多是两个异构体的比例不同导致这种结果的吗? 注:进口样品杂峰少,isolongifolenlactone 几乎没有 国产样品isolongifolenlactone 很多
液相色谱仪如何测定最大吸收波长
液相色谱仪如何测定最大吸收波长
新买的5110,用吸完超纯水的枪头去吸波长校准液,是否可能会把水带进去?如果带进去的话是否会把波长校准液稀释,从而影响仪器的测试结果?另外在仪器自动运行检测时(可打印检测报告)为何有的元素的强度并没有处于峰顶,而是在边上?
液相测多组分时,检测波长是分别取最大吸收波长测,还是建立一个共用的波长,同时测。如果分别测时,不能完全避免另外一个物质出峰,则含量怎么算。谢谢
关于分析物的最大吸收波长和在液相色谱上的保留问题:最近遇到一个比较郁闷的问题:PDA混标全扫描时,有一物质的最大吸收波长是225,保留时间是6.8min.在我做实际样品的时候,保留时间基本可以对上(相差0.02min),但是最大吸收波长却变成241,不知是何原因。。?望高手指教。混标用的是20%甲醇水溶液溶的,实际样品也是用20%甲醇水溶液,进样量和梯度都一样。
我要用比色法测定聚乙烯醇溶液的浓度,现在不知道波长应该选择在什么位置!具体分析方法是这样的:用移液管吸取聚乙烯醇试样10ml于50ml容量瓶中,稀释到刻度,然后取两个50ml容量瓶,一个加稀释的试样1ml,另一个加未稀释的试样,再分别加入1ml磷酸,0.02ml显色液摇匀,稀释到刻度。进行比色。(无稀释试样为参比)显色液:准确称取碘12.5克,碘化钾75克,硼砂10克,加水溶解并稀释到500ml即可。请问,比色时波长应该选择多少nm?谢谢!
[color=#444444]新和成了一种物质,想要液相色谱分析一下纯度,但不知道最大吸收波长,用紫外分光光度计做波长扫描,在330nm有最大吸收,可以在这个波长下用液相色谱分析吗?我也不知道这样做对不对[/color]
[color=#444444]用waters e2695液相色谱进行手性药物拆分,在取定波长范围内,改变波长时, 发现一个对映体的峰面积迅速增加,而另一个则没那么明显, 请问一下液相大神们 波长变化时手性对映体的吸收强度不是按浓度比改变的吗?[/color]
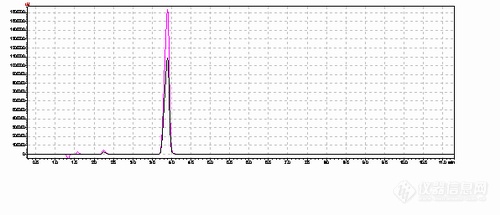
原料 乙酸长叶酯 进了仪器后 好多个峰 不知哪个是大家是如何定性的???附上图
过程分析仪器市场复杂,应用领域特定,市场增长受一些外部因素的影响。除在生产流水线上用于过程控制的分析仪器外,过程分析仪器还包括批量测定、现场环境测定的便携式分析仪以及运输、边境安检部门用于安全监测的化学、生物及核探测追踪仪。根据行业分析人员的观察,虽然在线分析仪器在安检和环境部门的应用保持了良好的增长势头,但在制药和化工行业的应用情况仍不令人如意,未达到5年前预期的水平。这是因为尽管压力是来自美国食品药品管理局,但适用于工业的培训计划需要由规章制定机构和政府部门制定,而大量的技术开发资金则需要制造企业投入。ABB Ltd (Zurich,Switzerland)是全球最大的专业在线分析仪器供应商,通过其“过程分析解决方案”每年向各类制造企业销售分析仪器2.5-3亿美元;其次为Emerson Electric Co(St Louis,MO)的过程技术部门,其年销售额约为2亿美元;Siemens AG(Munich,Germany)、Yokogawa Electric Corp(Tokyo,Japan)及Sick Maihak Inc(Minneapolis,MN)三家公司以1.5-2亿美元的年销售额紧随其后。其它重要企业还包括年销售额约为1亿美元的Ametek Inc(Paoli,PA)以及年销售额约为0.6-0.8亿美元的Endress&Hauser Group(Reinach,Switzerland)和Honeywell International Inc下属的自动化控制系统集团(Morristown,NJ)。最近的一份研究报告估计:2005年过程分析仪器的世界市场规模约为50亿美元,其中包括了操作成本、维修和综合服务费用。2002年这个数字是47.7亿,预计到2008年就将达到55亿美元。在Frost & Sullivan咨询机构去年一份名为“世界过程分析仪器市场”的报告中,估计过程分析仪器2004年仅仪器销售额一项就约22.8亿美元,并预计到2011年这个数值将突破30亿,年增长率约为4%,这个增长率略低于PAI Partners(Leonia,NJ)三年前一份报告[见Instrumenta 20(17)4]中的4.5%的复合增长率的预计。当时报告认为2003年在线分析仪器市场规模在14.2亿美元。然而以上数据也是有争议的。2005年夏戴安公司成立了过程分析仪器中心[见Instrumenta 22(6/7)15],过程分析中心经理Rich Cooley(原Eli Lilly经理)告诉Instrumenta,随着美国食品药品管理局过程分析技术创新行动计划(PAT)的公布[见Instrumenta 20(11)7],预计在制药和食品行业中过程分析仪器的市场份额将快速增长。根据Cooley的观点,过程分析仪器市场相当巨大而且在各领域应用都在增长,但是需要质疑的是所引用的数据的准确度如何,增长速度是否如市场研究报告所述那样快。Cooley认为至少对制药行业的情况估计过高,尽管市场具有很大潜力,但大多数行业对在线分析仪器的价值认识不足。与Cooley持有相同观点的人并不在少数,去年Control Magazine 的高级技术编辑Rich Merritt 在一篇文章中发表如下评论:大量最新上市的在线过程分析仪器并未提及符合PAT标准的要求。在Merritt看来,制造商似乎对于过程分析中心(CPAC Seattle,WA)的新型取样/传感器的创新[NeSSi,见Instrumenta 19(14)4]闭口不谈。CPAC从2000年就开始寻求推进与生产线一体化的标准化平台的过程分析仪器的小型化和模块化的发展。然而并不是所有的制造企业都对PAT和NeSSi持拒绝态度,还是有大多数的大型制药和生物技术企业已经投入人力专注于实施这两个标准。但是承诺实施并不等于许诺购买过程分析仪器。Cooley说,“PAT initiative实施5年了,虽然制药行业在此方面也有所行动,但是行动比较缓慢。因为制药行业具有谨慎的企业文化,所以人们对此并不感到吃惊。对于新技术的采纳,法律法规是一个很强的推动力,但是PAT并不是法律法规只是一种纯粹自愿的行为。过程分析仪器在环境监测领域的较快增长是因为在此领域法规执行严格并且力度大,在运输行业的安检因为有政府规章的强制,所以发展也很不错。”Cooley总结,“归根到底,虽然法律法规重要,但是财务利益才是关键。市场的真正增长来自于企业认识到在线分析仪器给他们带来经济利益,最大的挑战是要使企业认识到将分析仪器移至生产线的价值。对于制药行业来说,这是一个巨大的范式转变。我想对于我们来说最大的挑战是转变行业文化观念。”Cooley指出,“应对这种变化,制造商将要做的是开发出比当前方法有明显优势的新技术。技术的成本、仪器的简单化和易于维护对于企业来说是至关重要的,而现在的技术在以上方面仍然存在不足,这阻碍了行业的发展。但也表明企业有更大的机会通过技术创新促进市场的增长。分析仪器的小型化即是方向之一,并且在一定程度上使用者也在期待这些新的技术。”Steve Walton(PAI创始人之一,同时也是公司过程分析部门的主要分析专家)告诉Instrumenta :“关于PAT已经谈论很多了,但是人们没有意识到PAT是关于过程控制,分析仪器并不是必须要求的。基于这个原因,分析仪器的需求才没有厂商们预期的那样强烈,同时终端用户也还在观望。与过程分析仪器在制药行业的应用市场发展缓慢相比,NeSSi取得的成绩比较大,企业在技术方面的研究工作使他们认识到增加应用在线分析仪器会带来经济利益。当然与石化行业的巨大市场相比,制药行业的市场相对较小。”Walton指出,“然而,这种情况也并非出乎意料。对于制药行业这个保守的市场,新技术的推广需花很长的时间,但经济利益最终将促进新技术的推广。”新技术可能改变在线分析仪器市场的发展速度吗?Walton认为,“仪器供应商并不能比现在更快地促进市场发展,培训才是市场增长的唯一重要因素。”
问题: 各路大神一般打液相紫外,但是样品紫外吸收比较弱,用什么波长?[悠闲]回复: 扫DAD找到特征吸收波长,问题: 安捷伦1260紫外检测器可以走2个波长么回复: 可以中间切换,不能同时设置
我用的是热电M-5[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]仪,其他元素都可正常测定,但测定钾时,仪器提示波长找不到(能量太低),换为次灵敏波长也不行,换了空心阴极灯也不行(灯正常发光),望专家指点。谢谢!
液相波长的选择!!! 方法一:波长扫描,找最大吸收不就行了,一般是扫描,取最大波长。 办法二:如果测有关物质,一般选择254。如果测含量,一般选择最大吸收波长。 方法三:一般的液相上具有二极管阵列检测器的,都可以进行波长扫描,以取得最大吸收峰,如果没有这种功能的,可以从254开始,每格5或10的波长进行增加寻找,可以比较麻烦,也是没办法的办法.当然了,最好要有纯物质,这样比较准确点. 方法选择问题:比如,流动相配制\流速\取样量\波长\溶剂选择,以及检测器的选择等,都是要考虑的问题. 如果没有那种功能,可以用紫外扫描仪扫描,这样就可以找到最大吸收波长, 做波长扫描是最简单的方法,[col
液相波长的选择方法一:波长扫描,找最大吸收不就行了,一般是扫描,取最大波长。办法二:如果测有关物质,一般选择254。如果测含量,一般选择最大吸收波长。方法三:一般的液相上具有二极管阵列检测器的,都可以进行波长扫描,以取得最大吸收峰,如果没有这种功能的,可以从254开始,每格5或10的波长进行增加寻找,可以比较麻烦,也是没办法的办法.当然了,最好要有纯物质,这样比较准确点.方法选择问题:比如,流动相配制\流速\取样量\波长\溶剂选择,以及检测器的选择等,都是要考虑的问题.如果没有那种功能,可以用紫外扫描仪扫描,这样就可以找到最大吸收波长,做波长扫描是最简单的方法,如果不成,可以判断样品中的特定官能团,查相关文献,会有对此官能团的最大吸收波长,以其做参考,在其波长附近.做几个相差5或10nm的..
样品处理时用国药集团的95乙醇(050608)处理的,对照用色谱级乙醇配制的。因含量低复测,乙醇不够,换了另一个厂家的(020930),结果比原来的高2个百分点,排除对照误差,排除系统误差(重现性很好),称量也在范围内,只有溶剂有差别,紫外扫了一下发现确实有差别,050608的乙醇247nm吸收值为0.1多点,020930的乙醇吸收值在0.05,跟色谱级的乙醇差不多。而且200-300nm的出峰情况也不一样。020930的乙醇跟色谱级的乙醇差不多。我的疑问是1,溶剂有吸收对样品的影响应该是叠加还是消减?2,是溶剂在工作波长下有吸收,还是是溶剂本身带入的杂质对样品影响的?望各位大侠帮我解解惑吧!!!
各位大神,是否有直读光谱仪测量锌合金、锡合金的呢?诚请不吝赐教,指点下锌合金、锡合金的元素、通道、波长、分析曲线。谢谢各位,也替要买仪器的朋友谢过了。
液相色谱光谱分析中波长的几个峰为固定值,光谱分析在没有进行采样时即可进行,出来几个固定峰值,有何作用?而波长扫描时某个产品波长如何选择,与光谱分析的固定峰是否存在关系,如存在,有何关系
液相分析时,一般是选主成分和杂质的等比吸收波长作为分析波长吗?
方法一:波长扫描,找最大吸收不就行了,一般是扫描,取最大波长。 办法二:如果测有关物质,一般选择254。如果测含量,一般选择最大吸收波长。 方法三:一般的液相上具有二极管阵列检测器的,都可以进行波长扫描,以取得最大吸收峰,如果没有这种功能的,可以从254开始,每格5或10的波长进行增加寻找,可以比较麻烦,也是没办法的办法.当然了,最好要有纯物质,这样比较准确点. 方法选择问题:比如,流动相配制\流速\取样量\波长\溶剂选择,以及检测器的选择等,都是要考虑的问题. 如果没有那种功能,可以用紫外扫描仪扫描,这样就可以找到最大吸收波长, 做波长扫描是最简单的方法,如果不成,可以判断样品中的特定官能团,查相关文献,会有对此官能团的最大吸收波长,以其做参考,在其波长附近.做几个相差5或10nm的..
[align=center]液相面积归一法如何确定波长[/align]摘要:一般现在液相检测通常有面积归一法、校正面积归一法、外标法、内标法等几种方法,而检测纯物质,一般只采用面积归一法,而在确定杂质并提取到杂质对照品或提取到主成分对照品时,也可采用加校正因子或不加校正因子的主成分自身对照法进行检测。但是在实际检测过程中,有时候会出现波长确定比较难的情况,本文就如何在使用面积归一法时确定波长并且在主成分波长和杂质波长不同时如何进行选择提出可行性方案。 在使用液相进行检测一种物质的时候,如果没有标准方法、文献可供参考,我们需要通过以下几种方法进行判定波长,第一种是使用DAD二极管阵列检测器,可以很方便的确定被测组分,包括主成分和相关物质的最大吸收波长或者特征波长;第二种是通过紫外可见分光光度计进行波长的扫描,也能得到被测主成分的最大吸收波长或者特征波长,但是无法判定相关杂质的最大吸收波长,而且液相并不一定使用的是最大吸收波长,还可能从分离度等方面进行考虑。第三种是液相紫外检测器有波长扫描功能的,通过扫描波长进行确定目标组分的吸收波长,缺点仍然是只能一个波长测试一遍,才能进行分离度等其他参数的考察。 一般面积归一法测试进行选择波长的过程中,优先选择被测组分的最大吸收波长,其次是特征波长,并且要确定流动相在该波长的吸收不对被测成分产生干扰,选择波长最好比溶剂波长高20nm左右,这样干扰就比较小。但是单从面积归一化法这一个检测方法进行考虑,它是无法判断样品中相应成分的准确含量的。如果合成的相关化合物结构比较类似,都有紫外吸收,其紫外吸收曲线比较一致,有一定的参考价值。但一般情况下,其可靠性是很差的。但是,对于纯物质的判定,往往不能通过外标和内标法进行测定,而是采用面积归一法结合其他方法进行测定。如果出现纯物质的检测,主成分和相关杂质的在一个波长上响应不同,例如:该样品主成分在213和261nm处有两个吸收峰,213nm处为最大吸收峰。213nm测得的纯度较261nm低2%左右,杂质的出峰情况不同。紫外谱图如下:红色为213nm谱图,黑色为261nm谱图[img=,500,215]https://ng1.17img.cn/bbsfiles/images/2019/10/201910240958105464_2581_3295053_3.png!w500x215.jpg[/img][img=,500,312]https://ng1.17img.cn/bbsfiles/images/2019/10/201910240958108954_5224_3295053_3.png!w500x312.jpg[/img] 我们首先可以对主成分及主要杂质进行质谱分析,确定其化学式及其化学性质,如果有该杂质的对照品或者该杂质能够被提取出来,我们可以做杂质与主成分的校正因子,那么在某个波长下,该杂质的相对于主成分的校正因子满足0.8-1.2的范围内,我们就可以确认这个波长是适合的。 如果遇到比较难提取的杂质,但是主成分有相应的对照品,也可以在不同波长下进行主成分对照法进行,配制对照溶液并调节仪器灵敏度后,取供试品溶液和对照溶液适量,分别进样,测量供试品溶液色谱图上各杂质的峰面积,并与对照溶液主成分的峰面积比较,计算杂质含量。通过不同波长下面积归一法与主成分对照法测试的杂质含量的比值进行确定最佳检测波长。如果主成分也没有相应的对照品,这时可以采用衍生化法,不过这种方法仍需了解主成分及相关杂质的化学性质等,然后采用合适的衍生剂,将主要杂质和主成分用衍生的手段把吸收波长调整至非常接近,然后采用相同波长进行检测。这种需要的专业知识较高,而且衍生化法在实际应用中也存在衍生剂干扰、衍生不完全等情况。 还有一种方法是在通过NMR的氢谱、碳谱等相关的谱图先基本确定样品很纯的情况下,通过于其他检测器或者检测方法进行比对,比如我们可以通过卡尔费休法检测水分、顶空色谱检测残留溶剂、灼烧测定灰分、[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]测定一些小分子有机酸,然后结合紫外分光光度计、化学滴定等方法进行主成分的检测,并且与其在液相色谱上的各相关波长结果进行比对,即可得出仪器检测的最佳波长。综上所述,面积归一法并不能只看一个波长的结果,为了得到可靠的结果,我们需要综合判定其在不同波长下的结果进行比对,并根据主成分和杂质的波长响应,得到最佳检测波长,有条件的情况下,还需要通过其他检测器和检测方法进行比较。

正相色谱法采用极性固定相,流动相为相对非极性的疏水性溶剂,常加入一些极性溶剂以调节组分的保留时间,常用于分离中等极性和极性较强的化合物。反相色谱法一般用非极性固定相,流动相为水或缓冲液,常加入与水互溶的有机溶剂以调节保留时间,适用于分离非极性和极性较弱的化合物。对于某些易离解化合物的分析,为控制样品在分析过程的离解,常加入缓冲液控制流动相的pH值,增加保留,以改善分离。http://ng1.17img.cn/bbsfiles/images/2015/03/201503311236_540204_2960432_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/03/201503311237_540205_2960432_3.png根据以上列举的液相色谱的分析方法,在日常的分析检测中,我们都是采用了哪一类分析方法:注明检测的样品,仪器条件,分析条件。对于在检测中总结的经验,采取的措施也可以分享一下,以便于共同学习,共同进步。================汇======总===============1.看图解读畅谈之一:流动相与极性及PH值http://bbs.instrument.com.cn/shtml/20150304/5693051/2.看图解读畅谈之二:流动相与吸光度http://bbs.instrument.com.cn/shtml/20150304/5693078/3.看图解读畅谈之三:流动相与水http://bbs.instrument.com.cn/shtml/20150307/5698035/4.看图解读畅谈之四:流动相与粘度及分配比Khttp://bbs.instrument.com.cn/shtml/20150312/5705589/5.看图解读畅谈之五:色谱柱填料硅胶键合http://bbs.instrument.com.cn/shtml/20150323/5721015/6.看图解读畅谈之六:正反相液相色谱分析法——实例列举经验畅谈http://bbs.instrument.com.cn/shtml/20150331/5730704/
我们在做液相检测的时候一般设置的波长应该都是惨比波长吧?我想请问下对于未知物这个惨比波长一般是怎么确定的 是不是用紫外可见分光光度计进行全扫描然后根据最大吸收峰位置设定。 还有这个位置的最大吸收峰的波长和这个惨比波长有什么关系吗? 怎么才能确定最佳的惨比波长
最近发现,酸度不一样,扫描出来的最大吸收波长也不一样,我用的4.8-5.8的HAc-NaAc缓冲溶液,pH4.8-5.2的最大吸收波长基本在560-570,随着pH增大,有580、590、595nm,这正常么?如果不是在同一个波长下测A,我没法考察酸度对体系的影响
随着欧盟ROHS指令实施日期的日益临近,国内越来越多的相关企业在积极的思考和寻找应对的方案;X荧光分析技术(XRF)作为一种方便有效的快速分析手段,正迅速被业内人士所了解和应用。目前在中国市场上,应用于ROSH指令的X荧光分析仪均为能量色散类型;一般情况下,波长色散类型的X荧光分析仪器的准确度比能量色散类型的仪器要高很多;但应用于ROHS指令的场合时,波长色散和能量色散则各有优缺点,测量对象各有侧重;以下将从几个方面对两种类型的仪器进行比较和说明: 波长色散 能量色散 (Si-Pin)型 (SDD)型测量精度 20~50ppm 200~300ppm 100~200ppm测量时间 1~2分钟 4~6分钟 3~5分钟被测样品要求 规则形状需要制样 可以不规则形状 可以不规则形状 最佳应用范围 原料半成品成品电子元器件原料半成品成品电子元器件能量分辨率高,约15eV较低,约160eV较低,约160eV荧光强度高低较高技术复杂程度复杂简单较复杂使用寿命>10年>5年>5年仪器价格46万(国产)25万左右(国产)60万左右(进口)50万(国产)1、测量精度:尽管目前各家能量色散仪器(均为Si-Pin类型)生产商和销售商都给出了很高的技术指标,但在实际应用中(特别在被测样品不进行处理的情况下),真正可以期待的准确度都在200~300ppm之间(测量塑料中有害元素时,准确度会好一些;对不规则样品,则精度会更差);同时,对于同类型的仪器,进口仪器的指标和国产仪器之间并没有本质差别(基本配置),但进口仪器的价格却昂贵很多。波长色散X荧光分析仪的测量准确度比能量色散类型高一个数量级,基本在20~50ppm左右。2、测量时间:由于波长色散配备较大功率的X光管,荧光强度高;因此,波长色散仪器占用较短的测量时间,便能达到较高的测量精度。3、被测量样品的要求:由于技术特点的差异,波长色散X荧光分析仪需要对被测量样品进行简单的处理;对固体样品的一般处理方法是将被测量样品表面打磨光滑,对粉末和其他样品可以采用磨细后进行粉末压片法处理,相应的设备市场上很容易找到。能量色散型仪器最大的优势在于:可以对样品不作处理直接进行测量,对样品也没有任何损坏,直接用于生产的过程控制中;但需要强调指出的是:从荧光理论上讲,被测量样品的预先处理是必须的,对于能量色散仪器来说,我们可以采取一些技术手段进行校正来满足实际生产控制的需要,但即使采用了技术校正的手段,对不规则样品的直接测量也是以牺牲测量准确度作为代价的。4、最佳应用范围:由于波长色散和能量色散类型X荧光分析仪各自的技术特点,两种类型仪器所侧重的应用方案也不尽相同;波长色散X荧光分析仪具有较高的测量精度,但同时需要对被测量样品进行简单处理,更适用于进厂原材料、半成品、成品的精确检测和质量控制;能量色散X荧光分析仪虽然测量精度稍差,但具有快速、直接测量各种形状样品的优点,因此可直接在生产线上用于各种部件、电子元器件的检测。5、能量分辨率:能量分辨率是X荧光分析仪器的主要指标,分辨率数值越小,分辨率越高,仪器性能越好。6、荧光强度:对于X荧光分析仪器来说,各元素含量与该元素的荧光强度成正比关系;荧光强度越高,则统计误差越小,测量的准确度越高,仪器性能越好。7、使用寿命:波长色散类型仪器的使用寿命一般为10年以上;能量色散类型仪器的使用寿命一般也大于5年,影响能量色散型仪器寿命的主要因素是探测器部分的老化导致其性能指标变差。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=48263]波长色散X荧光分析仪与能量色散X荧光分析仪[/url]
符合比尔定律的有色溶液稀释时,其最大吸收峰的波长位置有变化吗?
各位大佬好,我刚入行,现在是用安捷伦高效液相色谱仪来检测样品,主要是看峰面积百分比来检测药品纯度。现在陷入了困惑。我没有准确的仪器来确认药品的紫外吸收波长,然后发现不同波长下药品的峰面积百分比含量都不同,那么怎样确定哪一个结果才是最准确的呢?
高效液相色谱用检测波长测定时一般都选择在对样品有最大吸收的波长下进行,以获得最大的灵敏度和抗干扰能力。但应特别注意在选择测定波长时,必须考虑到所使用的流动相的紫外吸收性质。也就是说,使用紫外-可见光检测器时,溶剂不应吸收测定波长的紫外光,样品测定波长应当在溶剂紫外吸收波长上限以上。噪声降至10-4~10-5AU,才能保证检测的灵敏度,才能用于梯度洗脱。液相色谱仪使用溶剂吸收波长的上限,就是透过波长的下限。波长的下限规定为溶剂在以空气为参比,样品池厚度为1厘米的条件下,恰好产生1.0吸光度时相对应的波长值,即溶剂透过率为10%时的波长。溶剂中如果含有吸收紫外光的杂质,同样会使检测背景提高,灵敏度降低,且用作梯度洗脱时会引起严重漂移。因此,鲁创分析认为液相色谱仪系统对溶剂纯度要求较高,一般应使用分光纯或分析纯溶剂,在有条件时,色谱纯溶剂为首选。应注意不能使用化学纯及纯度更低的溶剂。有时需要对溶剂进行专门纯化处理。
请教各位大侠,本人需要建立一个紫外的总含量测定方法,现扫描的全波长图如下。 吸收值大一点的为药材的吸收图,小一点的为标准品的图。 由于加入的显色剂非常大量,造成200-400段的吸收值波动非常大,但是均在427nm处有一个吸收峰。 唯一不同的是,药材在479nm处也有一个吸收峰,而标准品没有。现在想选择427nm作为一个检测波长。但是有同学说,此峰峰形并不是太好,也不对称,是一个尖的峰。所以求助各位大侠,帮忙看一下,这种情况,能不能选择这个峰作为检测波长,如果不能,有没有什么方法调整峰形。注:药材和标准品在400-700段是没有吸收的,在紫外下是有吸收。







 我要推广仪器
我要推广仪器
 下载APP
下载APP