使用varian GC450气相,PFPD检测器,新VF1701、30*0.25*0.25,色谱柱柱流量2.0ml/分钟,程序升温:80度保持1分钟,20度/分钟升到180度,5度/分钟升到230度,15度/分钟升到250度,保持7分钟,共24.33分钟。磷胺1在12.5分钟出峰,磷胺2在14.06分钟出峰,甲基对硫磷在14.10分钟出峰,这样磷胺2与甲基对硫磷完全重合。请高手指点,如何用VF1701分离磷胺与甲基对硫磷?

根据本人近期实验室经历,塑料中有机锡检出率似乎还不低,尤其是PVC类的样品。检出的物质,主要是单、二、三甲基锡,这与以往纺织品中易检出丁基锡、辛基锡的情况存在较显著不同。查阅了相关的资料,了解到目前有一种称为甲基硫醇锡的物质,在塑料行业被作为稳定剂大量应用,猜测这应该就是塑料中有机锡的一个主要来源。http://ng1.17img.cn/bbsfiles/images/2014/05/201405221403_500108_1846463_3.jpg欢迎大家发表看法。
自来水7月1日实行新国标,特把自来水中草甘膦、胺甲基硫酸的检测方法拿来共同分享
毒死蜱与甲基对硫磷分不开,用的是DB1701(30*0.25*0.25),新柱子时能分开,保留时间相差0.1min,用一年后,两峰完全重合,有什么办法能分开吗?
液相色谱串联质谱测试甲基硫菌灵,进的标品,第一针甲基硫菌灵出峰还正常,多菌灵峰很小只有甲基硫菌灵峰面积的十分之一,第二针就几乎不出甲基硫菌灵的峰只出多菌灵峰,再进两针就只有多菌灵完全没有甲基硫菌灵的峰了。流动相水相中有0.1%的甲酸,会导致这种转化吗?有人遇到过类似问题吗?请各位高手帮忙分析下,谢谢!
我做亚甲基蓝分光光度法测硫化物不显色 GB/T16489-1996 用的N,N——二甲基对苯二胺现配的,硫酸铁铵也是现配的,乙酸锌—乙酸钠也是现配的, 硫化物标准是买的,浓度为146微克每毫升的
测定大气中多环芳烃时,利用内标六甲基苯,打算用异辛烷配制,但不知道其在异辛烷中的溶解度或者饱和度,哪问高手知道指教一下?有没有做过的,讲授一下在配备过程中的注意事项。多谢啦
刚接手这个项目,按国标步骤一步一步做标准曲线。“取7支 100mL 具塞比色管各加20mL 乙酸锌乙酸钠溶液,分别加入0 0.50 1.00 2.00 3.00 和 5.00 mL 硫化钠标准使用液,加水至约60mL,沿比色管壁缓慢加入10mL N N 二甲基对苯二胺溶液,立即密塞并缓慢倒转一次,加1mL 硫酸铁铵溶液,立即密塞并充分摇匀放置10min 后,用水稀释至标线摇匀,使用1cm 比色皿以水作参比,在波长665nm 处测量吸光度。”刚开始做用的硫化物国家标准物质,标准系列有的显红色,有的显蓝色,吸光度没有线性关系,后来在不加乙酸锌乙酸钠溶液的情况下,空白为红色,其他为蓝色,作出了很好的线性。但由于标准物质用完了,用的很久以前的硫化钠基准物质按国标配制硫化物标准溶液,做出来的标准系列全显红色。想请问大家,什么原因导致显红色,大家遇到过吗,我问以前做这个项目的那个人,她说她从来没有遇到这种情况。
请问其他实验室的甲基对硫磷的保留时间是多少?
在苹果中加敌敌畏、甲基对硫磷两种农药,加标量为0.12mg/kg,用NY761来处理,敌敌畏的回收率可达100%,甲基对硫磷的回收率只有50%,实测值为0.06,少一半,是加标出现问题还是前处理有问题?
甲基异硫磷用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]质SCAN扫描,得出的质谱图,使用nistms程序检索,名字与分子式都与甲基异硫磷不一样,但是离子对与国标是一样的,这是怎么回事?
关于竹笋中甲基对硫磷含量的测定,有什么可以检测标准吗?
在用固相微萃取[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测甲硫醚和二甲基三硫醚时,为什么二甲基三硫醚老是不出啊
今天做氯仿中甲基对硫磷~FPD检测器~配的5,10,15,20,25的浓度曲线……分流比10曲线线性很不好~为什么??难道必须用不分流低浓度进样嘛?
1701柱,一个是DB,一个是VF1701MS,尺寸是一样的,30*0.25*0.25,可是DB1701上甲基异柳磷与水胺硫磷能分开,相差1.0min,而VF1701上只相差0.1min,两者重合,大家遇见过这种情况吗?
我手边的甲基硫菌灵的前处理方法都不好用,请高手提供?好像甲基硫菌灵不很稳定,有的都用衍生成多菌灵来作,到底有没有更好的方法?[em06]
异柳磷和甲基异柳磷有啥区别?
甲基对硫磷分光曲线谁有,谢谢
甲基对硫磷用程序升温时出峰很好,它与19种有机磷一起出峰,用DB1701柱,可是只有它一个时用恒温出峰很难看,恒温是190度保持20min,其它条件一样,只是程序升温与恒温的区别会影响峰形与峰面积吗?

做甲基对硫磷时,用高惰性衬管,出两个分叉峰,这是为什么?[img=,690,280]http://ng1.17img.cn/bbsfiles/images/2017/10/201710092201_01_1645480_3.jpg[/img]
我在做硫化物的实验时,用的标准溶液,是直接到标样所买的硫化物标准溶液100mg/L,我直接稀释至10mg/L,这样可以吗?还是需要按照国标的方法用硫化钠配制?做曲线的时候,国标上要求用100ML的比色管,我用的是50ML的比色管,也没有加入乙酸锌乙酸钠,直接取了标液,然后加水到40ML左右,再加入5ML二甲基对苯二胺溶液,然后是1ML的硫酸铁铵溶液,然后加水稀释到50ML标线。不知道这样做算不算是对国标方法的一种偏离呢?
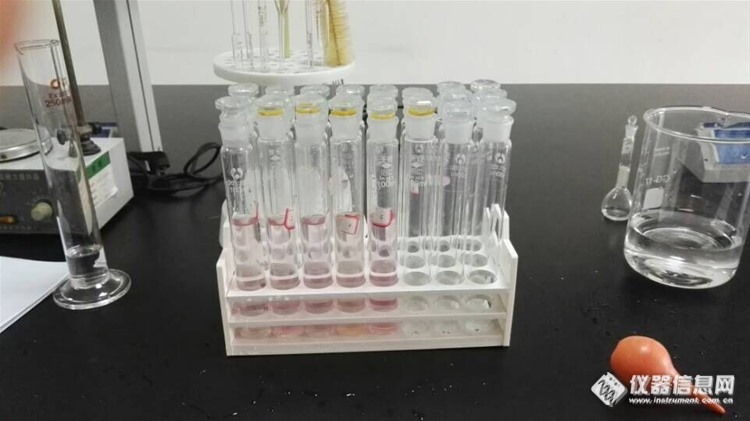
简单说下情况:由于我们采回来的水样用于很多项目的分析,所以硫化物是没有单独加固定剂保存的,对于一般地表水都是比较干净的,所以也没有进行预处理,具体操作按照国标做的:直接取50ml水样在比色管里,加20ml乙酸锌-乙酸钠,加水,加10ml对氨基二甲基苯胺,但是加上1ml硫酸铁氨以后,液体颜色变成红色,摇匀以后,静置10min,红色渐渐退去,之后就是基本没有什么颜色了,蓝色根本没有出现,这是什么原因?file:///C:\Users\fsd\Documents\Tencent Files\601551806\Image\C2C\8FBEA36AA6488770A3D5A705C559710D.jpghttp://ng1.17img.cn/bbsfiles/images/2016/06/201606032042_595973_3104264_3.jpg是应该将拿回来的水样取50ML加乙酸锌,加NaOH,沉淀过滤后取沉淀物于100ML比色管里,再加水,加其他试剂吗?沉淀物应该很少,又改怎么转移啊???感觉硫化物真的很难做啊,搞了很久都没搞得好。请各位老师解答下,谢谢。
在一个牛奶香精中分析出了甲基硫代磺酸甲酯(CAS:2949-92-0),也在GB2760里。我在想这个原料会不会是二甲基二硫醚氧化过来的?请各位大神解答!谢谢
求教:农残检测中对硫磷和甲基异硫磷如何分开,谁先出峰?安捷伦7890,进样口温度250,检测器250,柱温150(2分)然后6度每分钟的速率升到260(10分钟);DB-17色谱柱.
有没有谁参加了果蔬汁中马拉硫磷、甲基对硫磷残留量的测定?省局的实验室比对!请问结果多少?
硫酸锌可以和尿素发生反应吗?生成什么?再者我在看羧甲基纤维素钠的相关性质时也发现,它不能和重金属接触,请问下硫酸锌能和羧甲基纤维素钠发生反应吗?生成什么?
[color=#444444]各位大侠,我用FPD做马拉硫磷和甲基嘧啶磷,柱子是DB-1701的,但是两个峰,分不开,请求大家帮助[/color]
有人做过甲基硫菌灵在油菜上的残留实验吗?油菜的基质干扰比较大!!很恼火,大家有什么好的建议吗?
HJ 1226-2021 水质 硫化物的测定 亚甲基蓝分光光度法2021-12-16 发布 ,2022-03-01 实施该标准代替《水质 硫化物的测定 亚甲基蓝分光光度法》(GB/T 16489—1996),自改标准实施之日起,旧标准在相应的国家生态环境标准实施中停止执行。但实际情况,该标准生态部网页今天(2022-03-01)才刷新显示,一点准备都没有,被打个措手不及[img]https://simg.instrument.com.cn/bbs/images/default/em09504.gif[/img][img]https://simg.instrument.com.cn/bbs/images/default/em09509.gif[/img]
六甲基二硅胺烷和三甲基氯硅烷作为硅甲基化试剂与醇的反应机理! 另外,六甲基二硅胺烷可以与醇反应,为什么还要加入三甲基氯硅烷?三甲基氯硅烷不是也可以与醇反应吗?请指教!







 我要推广仪器
我要推广仪器
 下载APP
下载APP