[align=right][b]SGLC-GC-039[/b][/align][b]摘要:[/b]本文建立了砂仁中乙酸龙脑酯含量测定的GC 方法。结果表明,参照2020版《中国药典》中色谱条件并对升温程序进行优化,采用色谱柱SH-1 分析砂仁中乙酸龙脑酯,乙酸龙脑酯峰形对称,理论塔板数按乙酸龙脑酯峰计算远高于10000,满足《中国药典》要求。此方法可为砂仁中乙酸龙脑酯含量测定提供参考。。[b]关键词:[/b]砂仁 乙酸龙脑酯 SH-1 GC[b]1. 实验部分1.1 实验仪器及耗材[/b]Shimadzu GC-2030[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url];色谱柱:SH-50(30 m,0.25 mm × 0.25 μm;P/N:227-36162-01;S/N:1553669);SHIMSEN Arc Disc HPTFE针式过滤器(P/N:380-00341-05);[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]认证样品瓶LabTotal Vial(P/N:227-34002-01);SHIMSEN Pipet[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url]:SHIMSEN Pipet PMII-10(P/N:380-00751-02);SHIMSEN Pipet PMII-100(P/N:380-00751-04);SHIMSEN Pipet PMII-1000(P/N:380-00751-06)。[b]1.2 对照品溶液的制备[/b]取芳樟醇对照品适量,精密称定,加乙酸乙酯制成每1 mL含0.1 mg的溶液,即得。[b]1.3 供试品溶液的制备[/b]取本品粉末(过三号筛)约1 g,精密称定,置具塞锥形瓶中,精密加入无水乙醇25 mL,密塞,称定重量,超声处理(功率300W,频率40 kHz)30 分钟,放冷,用无水乙醇补足减失的重量,摇匀,滤过,取续滤液,即得。。[b]1.4 分析条件[/b]色谱柱:SH-1 (30 m, 0.25 mm × 0.25 μm P/N:221-75719-30;S/N:1541069 )升温程序:初始温度80 ℃,保持1分钟,以每分钟2 ℃的速率升温至100 ℃,保持5分钟载气:N2进样口温度:230 ℃分流模式:分流(10:1)控制模式:恒线速度(30 cm/s)初始流速:1.09 mL/min检测器:FID,温度:250 ℃进样量:1 μL[b]2. 实验结果[/b]按照上述色谱条件(1.4)进行采集,对照品溶液和供试品溶液色谱图如下:[b]对照品溶液[/b][img=砂仁中乙酸龙脑酯含量测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-GC-039_1.png[/img][font=arial, &][size=12px][/size][/font][b]供试品溶液[/b][img=砂仁中乙酸龙脑酯含量测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-GC-039_2.png[/img][font=arial, &][size=12px][/size][/font][b]重现性[/b][img=砂仁中乙酸龙脑酯含量测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-GC-039_3.png[/img][font=arial, &][size=12px][/size][/font][b]3. 结论[/b]本文建立了砂仁中乙酸龙脑酯含量测定的GC 方法。结果表明,参照2020版《中国药典》中色谱条件,采用色谱柱SH-1 分析砂仁中乙酸龙脑酯,乙酸龙脑酯峰形对称,理论塔板数按乙酸龙脑酯峰计算远高于10000,满足《中国药典》要求。此方法可为砂仁中乙酸龙脑酯含量测定提供参考。
告急:哪位知道新版本测定乙酸正丁酯的国标标准。多谢!
标准配法是吸取2ml乙酸正丁酯用60%乙醇定容到100ml,怎么从安瓿瓶中吸2ml出来?移液管都比安瓿瓶粗啊?
我在GDX-102气象色谱柱(热导池法)上测混合物(含乙酸。乙醇。正丙醇。异丁醇。异戊醇。乙酸乙酯。乙酸正丙酯。乙酸异丁酯。乙酸异戊酯),请问专家:怎样控制气象色谱的操作条件能把这些混合物各自的含量测出来,而且峰形较好
请问 GB/T22338--2008 中说, 样品的脂肪含量较高时,可以用三氯乙酸溶液饱和的正已烷液-液分配除脂后再用SPE柱净化。请问这一步具体怎么操作啊?还有三氯乙酸溶液饱和的正已烷又怎么配制啊?谢谢啊
用乙酸和丁醇反应,最后蒸馏的温度(124)上不去是啥原因急急!!
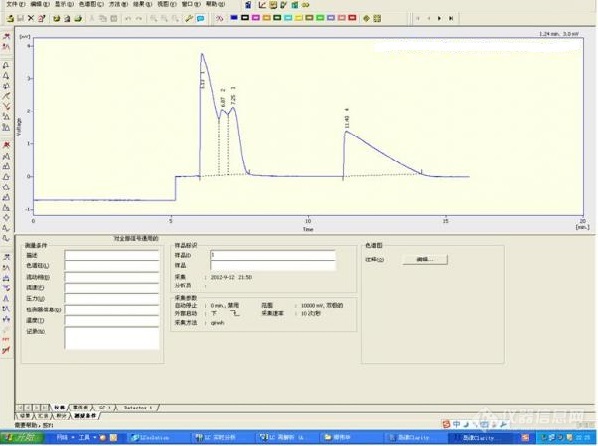
[color=#444444] 体系:[/color][color=#444444]乙酸/正丁醇/乙酸正丁酯/水;[/color][color=#444444]质量分数:均为25%[/color][color=#444444]沸点:乙酸119℃,正丁醇118℃,乙酸正丁酯126℃,水100℃。[/color][color=#444444] 分析条件:[/color][color=#444444]1. 色谱柱:DB-FFAP (30×0.25×0.25)[/color][color=#444444]2. 检测器:TCD[/color][color=#444444]载气流量:N2,0.25ml/min[/color][color=#444444]进样口温度:150℃[/color][color=#444444]柱温箱温度:120℃[/color][color=#444444]检测器温度:220℃[/color][color=#444444]图上可以看出:[/color][color=#444444]1, 基线发生瞬间跳动,一般基本上是漂移吧,为什么我这会突然发生阶跃式的跳动?分析原因是不是载气流量太低,可能载气流量发生不稳定,导致基线发生跳动?[/color][color=#444444]2, 由于有四元组分,前三个峰没有完全分开,而色谱柱分离效率跟载气流量,柱温关系最大,载气流量已经很低了,而且柱温也低于乙酸正丁酯的沸点,是不是也不能再低了。考虑能不能采用程序升温的方法,有没有可能将峰分开?如果可以的话,程序升温建议应该如何设置?[/color][color=#444444]求高手。[/color][color=#444444][img=,598,446]https://ng1.17img.cn/bbsfiles/images/2019/09/201909271014211801_6141_1752329_3.jpg!w598x446.jpg[/img][/color]
求助三氯吡氧乙酸、2,4-二氯苯氧乙酸正丁基酯(2,4-D丁酯)的检测方法
我们是白酒企业,我想请问一下,分析白酒中己酸乙酯所用内标:(乙酸正丁酯和乙酸正戊酯)的详细配制方法.
要分析乙酸中的甲酸和乙酸乙酯,TCD检测器,填充柱。 用GDX加酸不太好分离,请教诸位同仁有无经验?
各位大佬,毕业做乙酸正丁酯的有机合成,要求产率达到80%以上,纯度99.9以上,现在纯度终于做到了,就是产率一直在50%上下左右,跪求各位大佬,疫情原因拖了这么久,马上就六月份了,这可咋办 跪求大佬!!! 下面是我整理的合成细节,大家看看那里不对或者可以做得更好的地方,请大家指出,各位的意见对我很重要谢谢????[img=,690,1226]https://ng1.17img.cn/bbsfiles/images/2020/04/202004132232031655_3401_3490514_3.png[/img][img=,690,1226]https://ng1.17img.cn/bbsfiles/images/2020/04/202004132232037385_8371_3490514_3.png[/img]
用TENAX采样管进样做TVOC,为什么乙酸正丁酯出不来?求解?
我买的坛墨的7种饱和脂肪酸酯类化合物混标!甲酸甲酯 乙酯 乙酸甲乙丙丁戊酯。现在问题来了。我甲酸乙酯和乙酸甲酯一直分不开 。下面是我条件:30 度保持4min 以1 度/min 升到40度 30度/min到100。后面都没问题 甲酸甲酯我温度低一点也能和cs2分开。可是甲酸乙酯和乙酸甲酯...分不开。那我怎么做呢?是不是要单独做一个乙酸甲酯曲线方法验证。柱子是 ffap 30 0.32 0.25[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2020/02/202002211425479329_4319_2990176_3.png[/img]
用茚三酮法测定氨基酸总量的时候先要配制乙酸-乙酸钠缓冲液,实验指导派介绍的是先称45.5克乙酸纳溶于100ml水中,加热溶解蒸发至60ml,冷却后加30ml乙酸,在用水定容于100ml容量瓶中.但我们在具体操作中却怎么也做不到,先是加热够冷却,但发现烧杯壁上附着厚厚一层药品,并且容量瓶里的溶液很快变成了糨糊,30ml乙酸和10ml的水根本冲不干净,误差太大.第二次干脆在还温着的时候就倒进容量瓶,但效果还是和第一次差不多.第三次是加热后趁热加乙酸进去,但还是那样.不知道别人做的时候有没有这种情况,是怎么解决的?我急啊~~~~~~~~~~~~~~~~~~```[em16] [em49]
我想用填充柱分析乙酸酯类,有:乙醇、乙酸乙酯、乙酸甲酯、乙酸正丙酯、乙酸异丙酯、丙酸乙酯、乙酸正丁酯,该选择什么类型的填充柱(固定液,担体),要几米。因为我的仪器没有毛细管系统,只要填充柱系统,FID。谢谢了
用TENAX采样管进样做TVOC,为什么乙酸正丁酯出不来?求解?
给位大侠,我在根据国标法测奶粉中的牛磺酸时,配置乙酸锌溶液用来去蛋白,可是乙酸锌不溶于水。通过加热溶解时发现40℃时几乎没溶,后来水浴温度调到80℃时不停搅拌后还有一些未溶,但是温度再高的话里面的水又会蒸发了,于是又加了点凉水进去,随着温度的降低乙酸锌又被析出来了,放置一夜后烧杯上全是乙酸锌,而且烧杯里面乙酸锌又与原先加水时的量一样多,这是怎么回事?
最近没有事,常想一个问题,我们做金中杂质分析时用乙酸乙酯,而很多单位却用乙醚,大家能告诉我吗?我试着比较两种物质.对比如下1\安全性不用说乙醚危险,极易燃烧,属于管制药品.而乙酸乙酯安全的多,而且满大街都有卖的.2\操作乙醚在萃取过程 ,分层很慢,有的单位做试样时,需要静止5分钟以上,而乙酸乙酯只需要静止半分钟就完全能分离.3\效果乙醚萃取时,如果时间不够长,通常水相颜色很深(萃取不完全),而乙酸乙酯不出现这个问题.4\纯化 乙醚的沸点低,在34度就能进行蒸馏,而乙酸乙酯纯化稍微麻烦,但是在78度蒸馏产物完全能达到萃取效果,而且其中杂质含量和乙醚蒸馏后大致相当.5\金回收由于乙醚沸点较低,按理将其蒸发很容易,然而温度太高,乙醚易着火燃烧,而使金损失严重,另外乙醚直接排出也不利于环保,所以只能通过蒸馏来回收乙醚并分析金.但是在该过程中,蒸馏反应剧烈,可能造成喷溅.相比而言,乙酸乙酯由于沸点高,所以直接在电热板上加热除去乙酸乙酯即可.从上面我觉得应该选择乙酸乙酯,而大多数单位却选择了乙醚,这是为什么呢?
利用顶空[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定甲乙丙丁戊5种醇,分别测试了下正戊烷、正庚烷、丙酮、乙酸乙酯都能够与这5种醇分开,乙酸乙酯我已经选来做稀释剂,其他三种合适做内标物质吗?
[color=#444444]各位前辈,请问乙酸乙酯分析纯经蒸馏,其中间液可以作为流动相进液相吗?[/color][color=#444444]晚辈愚见:分析纯和色谱纯的区别并不在于杂质含量的高低(分析纯试剂的杂质也可能比色谱纯低),而是色谱纯试剂去掉了有紫外吸收的部分。但是我们实验室没有色谱纯的乙酸乙酯,所以想用其蒸馏之后的中间液,请问这样可以吗?[/color][color=#444444]请前辈们指教,灰常感谢。[/color]
乙酸乙酯沸点77.06℃,折光率1.372 3,相对密度0.9003。乙酸乙酯一般含量为95%~98%, 含有少量水、乙醇和乙酸。可用下法纯化:于1000mL乙酸乙酯中加入100mL乙酸酐,10滴浓硫酸,加热回流4h,除去乙醇和水等杂质,然后进行蒸馏。馏液用20~30g无水碳酸钾振荡,再蒸馏。产物沸点为77℃,纯度可达以上99%。
求助各位老师,关于乙酸乙酯的测定,样品百分比含量大约在90-98%左右,要求是使用内标标准曲线法,在建立曲线的时候浓度应该怎么选择?溶剂是乙醇标液和内标物需要自己配内标物是使用乙酸正丙酯。
大家好,我急求用岛津气相色谱分离,乙酸和乙酸乙酯的分离条件,柱子,进样口,检测器温度,以及乙醇和乙酸乙酯的分离条件,柱子采用RTX-17ms,急求,谢谢大家!!!
做偶氮二异丁腈,发现有资料用乙酸甲酯:水=1:1做流动相,反相色谱,所以想问问乙酸甲酯是否可以用于反相色谱?乙酸乙酯是用于正相色谱,所以求助
大家有做过这个巯基乙酸的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]验证吗,溶剂是乙腈:水=1:9。然后就发现空白也有值,就考虑试剂干扰,分析后发现乙腈没有影响,而水居然有干扰,想知道大家做的时候出现过这种情况吗,流动相为0.1%甲酸水和乙腈流动相。如果我把溶剂换成纯乙腈,巯基乙酸能溶吗,这会影响到样品的提取吗
各位大神,哪有有二正辛基-双(巯乙酸2-乙基己酯)(DOTE)和三(2-乙基己基巯基乙酸)辛锡(MOTE)反应物料,或者有卖三(2-乙基己基巯基乙酸)辛锡(MOTE)的供应商啊。网上百度,相关信息为零。这是SVHC中的一项物质。http://simg.instrument.com.cn/bbs/images/default/em09512.gif
各位同仁: 我使用的是北分sp3420气相色谱,分析条件是:90度保留8.5分钟,每分钟50度升至终温250度,保留2分钟。我进tvoc标样时乙酸丁酯不出峰是怎么回事啊
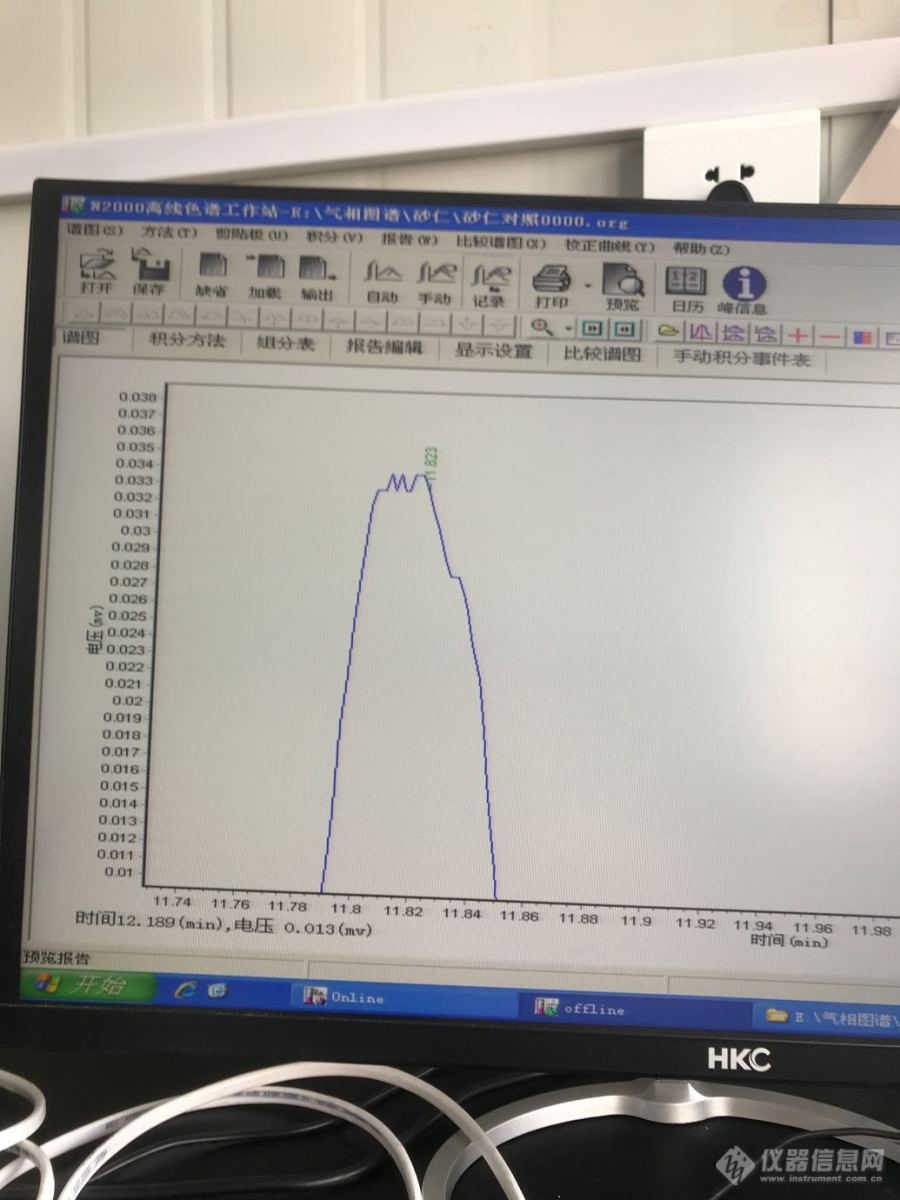
砂仁含量测定,乙酸龙脑脂对照品的峰是这样的,什么原因?[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/09/201909291048233492_354_4008962_3.png[/img][img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/09/201909291048362627_1375_4008962_3.png[/img][img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/09/201909291048546892_2814_4008962_3.png[/img]
之前用的tenax管所有峰都出,然后今天换了一根管子。活化了几个小时了,也没有杂峰了,结果进了2ul,其他峰全部出峰正常。就只有乙酸正丁酯不出峰。重复也是如此。[img]https://ng1.17img.cn/bbsfiles/images/2019/02/201902271657048417_9800_3519857_3.png[/img]
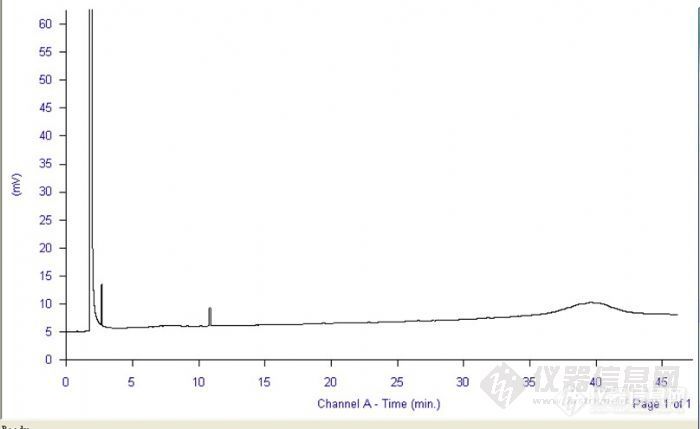
第一次试验做了个白酒分析方法中的乙酸乙酯,内标法用乙酸乙酯标样做了校正因子的图谱,实验条件是:N2流速为1.0ml/min,分流比10:1,柱温60度恒温3min,以3.5度速率升至180度,恒温10min。吸取2%乙酸乙酯1ml于100ml容量瓶中,加入内标2%乙酸戊酯溶液1ml,用乙醇稀释至刻度。测出图谱如下图由于是第一次做,没有图谱进行比较,不知出来的峰是否分的开,哪个又是哪个峰,有点糊涂,哪位同仁有按照GB/T10345-2007做过该图的,希望能分享一下图谱。为啥基线到40分钟左右出了个峰?http://ng1.17img.cn/bbsfiles/images/2012/06/201206041039_370210_2210929_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP