[size=4]各位高手: 有谁知道缩宫素的效价测定方法?流动相是什么组成和配比,以及效价计算方法 谢谢各位指导![/size]
[size=3]各位高手,有谁知道缩宫素效价的计算公式?跪求指导,谢谢![/size]
[size=4] 本人目前在做一个缩宫素标准的工作,需要2010版中国药典中有关缩宫素原料药或制剂标准作为参考。因为我们还没有条件看到2010版药典,希望好心的朋友能把2010版药典中关于缩宫素原料药或制剂的标准扫描或拍照提供给我,不胜感激。 邮箱:samesir@163.com[/size]
串联[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]做缩宫素,查不到相关资料,哪位老师可否分享一下相关资料,不胜感激。

【作者】 高岩; 席时东;【Author】 Gao Yan Xi Shidong (Ningbo Institute for Drug Control,Ningbo,Zhejiang,China 315040)【机构】 浙江省宁波市药品检验所; 浙江省宁波市药品检验所 浙江 宁波 315040; 浙江 宁波 315040;【摘要】 目的测定缩宫素的含量。方法采用高效液相色谱梯度洗脱法,选用Diamonsil C18柱,以15.6 g/mL磷酸二氢钠溶液及乙腈-水(1:1)为流动相梯度洗脱,检测波长为220 nm。结果线性范围为0.5~100 IU/mL。平均加样回收率为96.9%。结论高效液相色谱法所用方法准确、简便。 更多还原【Abstract】 Objective To establish a HPLC method for assaying of oxitocin.Methods The gradient elution was used with Diamonsil C18 column and UV detection at 220 nm.The mobile phase was 15.6g/mL sodium dihydrogen phosphate buffer and acetonitrile-water(1:1). Results The oxitocin curves were linear between 0.5-100 IU/mL(r=1.0000),and the recovery rate of the standards added was 96.9%. Condusion The method is accurate,simple. 更多还原【关键词】 高效液相色谱法; 缩宫素; 含量测定; 【Key words】 HPLC; oxitocin; content determination; http://ng1.17img.cn/bbsfiles/images/2012/08/201208271552_386451_2352694_3.jpg
近期,中科院地球化学研究所环境地球化学国家重点实验室冯新斌研究员带领研究团队在利用汞同位素示踪汞污染源研究方面取得新进展,为准确解析和评估环境流域中污染物的来源提供了有力的技术手段和理论依据。 汞是环境中毒性最强的重金属之一。环境汞污染问题一直是世界各国关注的焦点和热点。作为中国经济发达和城镇化建设最为典型的区域之一,珠江三角洲东江流域汞污染日益严重。准确分析环境流域中汞的来源和归趋问题不仅是目前研究汞的环境生物地球化学过程的难点,而且对评估和治理环境流域中汞污染具有重要意义。稳定同位素示踪是地球化学研究中的重要内容和技术。目前,国际上初步建立的汞同位素体系已明确汞同位素可以作为汞污染源和生物地球化学反应及其发生程度的示踪剂。 冯新斌研究员带领研究团队利用地化所矿床地球化学国家重点实验室的多接收电感耦合等离子体质谱仪(MC-ICP-MS),建立了一套精准的测定样品中汞同位素的方法,同时利用此项技术对东江流域沉积物汞同位素特征进行了深入的研究。研究结果表明,东江沉积物中不同生态单元的汞污染程度和汞同位素特征差异显著。通过深入分析和合理推断,结合沉积物中汞质量分馏和非质量分馏明显特征(图1),研究人员建立了流域汞污染源(自然源,生活源和工业源)三元混合模型,并采用东江流域各生态单元沉积物汞含量进行模型检验,明确证明不同来源的汞具有不同的汞同位素比值(图2)。由此证明,汞同位素技术可以有效用于示踪和量化沉积物中不同来源的汞。 相关研究成果已分别在地球化学和环境科学领域的国际杂志Chemical Geology(2011,287:81-89) 、(http://www.gdlord.com)Chinese Journal of Analytical Chemistry (2010, 38(7):929-934)、Applied Geochemistry(2010, 25:1467-1477) 等期刊上发表。 目前,冯新斌研究员带领的有害污染物研究课题组仍在进一步探索汞同位素技术在环境科学和地球化学领域的应用与发展。http://photocdn.sohu.com/20110922/Img320129065.jpg图1. 东江沉积物中不同生态单元的汞同位素特征(δ202Hg vs Δ199Hg)http://photocdn.sohu.com/20110922/Img320129068.jpg图2. 东江沉积物中不同汞来源的贡献比例(X, Y, Z 分别代表工业源,生活源和自然源)来源地球化学研究所)
锁阳咖啡中咖啡因含量的测定锁阳咖啡以天然锁阳为基础,配以速溶咖啡,是近几年涌现出的一种新型的固体饮料。锁阳主产于甘肃、青海、内蒙,其中以甘肃嘉酒地区的锁阳产量最大、质地最优。锁阳可调节生理机能、均衡营养、促进血液循环、滋肝健肾。锁阳咖啡是锁阳和咖啡的有机结合,越来越受到消费者的青睐。但是人们在饮用锁阳咖啡的同时却容易忽略咖啡的品质,咖啡因是咖啡中的一种重要成分,也是衡量其质量的一项重要指标,作为一种中枢兴奋剂,能兴奋大脑皮层,但易上瘾,因而国家对其制定了相应的标准。本实验用SN/T 1391-2004 《进出口速溶咖啡检验规程》中速溶咖啡中咖啡因的测定方法测定锁阳咖啡中咖啡因的含量。http://ng1.17img.cn/bbsfiles/images/2013/07/201307291536_454526_2764104_3.jpg1.方法提要样品用水溶解过滤后,用配有紫外检测器的高效液相色谱(HPLC)测定咖啡因,外标法定量。2.试剂和材料所有试剂除特殊注明外,均为分析纯,水为超纯水。2.1 乙睛:色谱纯;2.2 咖啡因标准品:纯度≥99%;2.3 标准储备液(100m g/L),准确称取 。0.0100 g 咖啡因标准品于100m L容量瓶中,用水溶解并定容至刻度,作为标准储备液。根据需要再用水将标准储备液稀释成适当浓度的标准工作液。3.仪器和设备3.1高效液相色谱仪配紫外检测器。3.2超声波振荡器。4.测定步骤4.1 提取称 取 0.1g(准确至 0.0001 g )均匀试样于 100m L容量瓶中,加人 80m L水,置超声波振荡器中超声20 min,冷却后用水定容至刻度并混匀,过0.45滤膜后,供HPLC测定。4.2 测定4 .2 .1 色谱条件a) 液 相色谱仪,配紫外检测器,检测波长273n m;b) 色 谱柱:C,。柱(25cm×4.6m mID,5um)柱或相当柱;c) 流 动相:乙睛一水一乙酸(16+83十1);d) 流 速 0.5 m L/min;e) 进 样 量 5uL4.2.2 色谱测定根据样液中咖啡因的含量情况,选定峰面积相近的标准工作溶液。标准工作溶液和样液中的咖啡因的响应值均应在仪器的检测线性范围内对标准工作溶液和样液等体积参插进样测定。在上述色谱条件下,咖啡因的保留时间约为9min。 http://ng1.17img.cn/bbsfiles/images/2013/07/201307291536_454528_2764104_3.jpg 标准溶液色谱图 http://ng1.17img.cn/bbsfiles/images/2013/07/201307291537_454529_2764104_3.jpg 标准曲线 http://ng1.17img.cn/bbsfiles/images/2013/07/201307291537_454530_2764104_3.jpg 样品色谱图 4.2.3[/size
我想知道荧光素钠和罗丹明6G的pKa,这样就可以知道它们在什么pH条件下呈中性。有哪位大虾知道,帮忙告诉一下!谢谢!
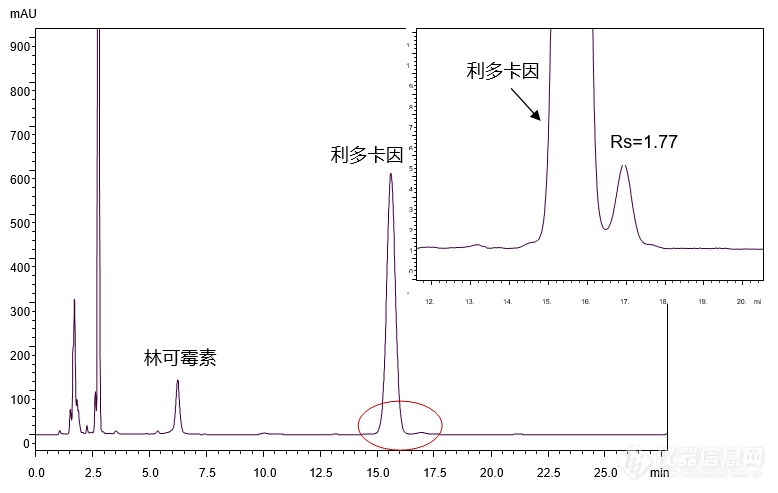
[align=center][b]【国家药品标准】林可霉素利多卡因凝胶的分析[/b][/align][align=center][b][/b][/align][align=right][b]——依据国家药品标准WS-10001-(HD-0140)-2002方法[/b][/align][b]林可霉素利多卡因凝胶[/b]为复方制剂,每克含林可霉素5毫克,利多卡因4毫克。适应症为用于轻度烧伤、创伤及蚊虫叮咬引起的各种皮肤感染。 [img=,193,127]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834522166_2994_2222981_3.gif!w193x127.jpg[/img] [img=,140,64]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834520028_3541_2222981_3.gif!w140x64.jpg[/img] 林可霉素 利多卡因 Lincomycin Lidocaine M.W.: 406.54 M.W.: 234.34客户提供林可霉素利多卡因凝胶样品,希望本实验室帮忙通过筛选色谱柱及调节分析条件,依据[color=#ff0000][b]国家药品标准WS-10001-(HD-0140)-2002[/b][/color]方法,实现林可霉素利多卡因凝胶样品的良好分析。首先,使用能在纯水条件下稳定使用的高极性色谱柱[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ S5 4.6 mm i.d. × 150 mm[/b][/color],对林可霉素利多卡因凝胶样品进行分析,结果如图1所示,[color=#330099]利多卡因与其峰后杂质之间分离度为1.77[/color]。[align=center][img=,690,437]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858200006_8607_2222981_3.png!w690x437.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18 [/sub]AQ分析所得色谱图[/align]注:峰上标数字为分离度。[img=,528,205]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858202566_2695_2222981_3.png!w528x205.jpg[/img]为进一步提高利多卡因与其峰后杂质之间的分离度,在原条件基础上将柱温由30℃降低至25℃,并分别使用 CAPCELL PAK C[sub]18[/sub] AQ、CAPCELL PAK C[sub]18[/sub] MG及高含碳量ODS色谱柱SUPERIOREX ODS进行分析,结果如图2所示。[align=center][img=,690,490]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859201516_7229_2222981_3.png!w690x490.jpg[/img][/align][align=center]图2 25℃条件下不同色谱柱分析结果对比[/align]注:峰上标数字为分离度。[img=,637,223]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859204236_7198_2222981_3.png!w637x223.jpg[/img]如图2所示,在柱温25℃条件下使用三款色谱柱进行分析,其中,[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ色谱柱分析结果最好,利多卡因与其峰后杂质分离得到最佳分离,分离度为4.23[/b][/color];[color=#330099][b]使用CAPCELL PAK C[sub]18[/sub] MG色谱柱进行分析时,利多卡因与其峰后杂质分离度为3.27[/b][/color];而使用SUPERIOREX ODS色谱柱分析时,利多卡因与其峰后杂质未得到有效分离。综上,在国家药品标准WS-10001-(HD-0140)-2002方法基础上,将色谱柱柱温由30℃降低至25℃,使用高极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ及中等极性色谱柱CAPCELL PAK C[sub]18[/sub] MG进行分析,均可在25 min内完成林可霉素利多卡因凝胶样品的分析,并得到利多卡因与其峰后杂质之间的良好分离结果。[align=right][/align][align=right][/align][align=right] [/align][align=right]三耀精细化工品销售(中国)有限公司[/align][align=right]技术开发部[/align][align=right]地址:北京经济技术开发区宏达南路5号[/align][align=right]宏达利德工业园1栋418室[/align][align=right]邮编:100176[/align]
同位素质谱现在用哪款空气压缩机啊?http://simg.instrument.com.cn/bbs/images/default/em0817.gif
http://v.t.qq.com/share/images/s/b16.png 求助] [b]尿素中缩二脲的测定[/b] 1.GB/T 2441.2-2010《尿素的测定方法 缩二脲含量 分光光度法》只适用于由氨和二氧化碳合成制得的尿素缩二脲含量。哪其他方法制的尿素如何测缩二脲?2.HG/T 2843-1997中缩二脲标液的配置是不是必须用氨水溶液洗涤缩二脲哪,买的缩二脲标准物质还要这样提纯吗? http://v.t.qq.com/share/images/s/b16.png 求助] [b]尿素中缩二脲的测定[/b] 1.GB/T 2441.2-2010《尿素的测定方法 缩二脲含量 分光光度法》只适用于由氨和二氧化碳合成制得的尿素缩二脲含量。哪其他方法制的尿素如何测缩二脲?2.HG/T 2843-1997中缩二脲标液的配置是不是必须用氨水溶液洗涤缩二脲哪,买的缩二脲标准物质还要这样提纯吗? [size=3]1.GB/T 2441.2-2010《尿素的测定方法 缩二脲含量 分光光度法》只适用于由氨和二氧化碳合成制得的尿素缩二脲含量。哪其他方法制的尿素如何测缩二脲?2.HG/T 2843-1997中缩二脲标液的配置是不是必须用氨水溶液洗涤缩二脲哪,买的缩二脲标准物质还要这样提纯吗?[/size]

作者:赵玉静;(沈阳市皇姑区中医院;)摘要:目的建立宫炎片的质量控制标准。方法采用HPLC法对丹参素钠进行含量测定。Diamonsil TMC185μ(250×4.6mm)色谱柱;甲醇-水-0.5%三氯乙酸(8∶90∶2)为流动相;流速0.8ml/min;柱温25℃;检测波长:280nm。结果HPLC定量方法分离效果好,线性范围0.202-1.01μg。平均加样回收率98.7%,RSD为2.3%。结论本法可作为宫炎片的质量控制方法。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208131452_383527_1606903_3.jpg
GBT 17739.3-2004 技术图样与技术文件的缩微摄影第3部分35 mm缩微胶片开窗卡.pdf
GB/T 17739.5-2006 技术图样与技术文件的缩微摄影 第5部分:开窗卡中缩微影像重氮复制的检验程序- .pdf
小白一枚,做维生素C钠和维生素C中的汞时遇到很神奇的事情,首先配制的是汞标,分别为0.5ppb、1ppb、1.5ppb,然后样品浓度为100mg/ml,用标准曲线法测定,线性很好,但是做回收率的时候,回收率能达到300%-400%。然后我又将维生素C消解了一下,重新做回收率,这回回收率达到75%。实在是想不明白为啥,请各位大神指点一下,小弟先在此谢过
它是采用免疫原理和胶体金层析技术制成快速检测牛奶、食品、饲料中的霉菌毒素的残留量检测限是10ppb 饲料 谷物用黄曲霉毒素总量检测卡,黄曲霉毒素B1检测卡检测限是0.5 ppb 原奶生鲜奶 黄曲霉毒素M1检测卡检测限是50ppb 饲料 谷物 玉米赤霉烯酮检测卡检测限是1ppb 饲料 谷物 赭曲霉毒素检测卡检测限100ppb 伏马毒素检测卡检测限0.3毫克每千克 液态奶、奶粉 三聚氰胺检测卡
大家好,有一个问题需要大家帮忙看看。我手头有一个汞样品,客户要求测定样品中的汞是哪个同位素,而且要知道含量或者百分比。样品里可能是一种汞的同位素,也可能是几种同位素的混合物,用哪种质谱来测比较合适,才能分开这几种同位素?另外就是给推荐一下测试的机构和高校和研究所,联系电话等等。谢谢。
般而言,人们都希望饮料澄清透明。比如茶,清亮的总比浑浊的更有吸引力一些。所以,人们发现有的红茶茶汤在放凉之后出现浅褐色或橙色乳状的浑浊之后,在相当长的时间里并不待见它。直到后来有农艺师指出这其实是优质红茶的标志,这种被称为“冷后浑”的现象才受到人们的欢迎。冷后浑是如何产生的?为什么它又被认为是好茶的标志呢?茶叶中有许多种成分,其中有一类在化学结构上有共同之处,统称为茶多酚,现在已经识别出了有几十种。在未经加工的茶叶中,茶多酚大多数以儿茶素的形态存在。红茶制作中要进行充分的氧化,许多儿茶素会转化成茶黄素,还有的会进一步转化成茶红素。茶黄素和茶红素,就是为红茶带来红亮颜色的功臣。茶黄素的溶解度受温度影响比较大。在高温下,它还能好好地呆在茶汤中。当温度降低,它们就开始扎堆。温度越低,扎的堆就越大。大到一定程度——大致相当于牛奶中的乳滴大小,看起来就是茶汤变浑浊了。再进一步扎堆,就会形成乳酪那样的东西,与茶汤分层。茶中还有一种成分是咖啡因。其实它跟茶黄素一样,随着温度的降低也会喜欢扎堆,溶解度也会降低。不过在茶汤中的咖啡因含量低于它的溶解度,所以它们自己并不足以导致茶汤浑浊。但咖啡因非常喜欢茶黄素——相对于自己扎堆,它们更喜欢跟茶黄素混在一起。一个茶黄素分子上有两个位置能够结合咖啡因,当第一个咖啡因分子傍上茶黄素之后,就会使茶黄素露出第二个结合位点,再容纳另一个咖啡因分子。咖啡因到了人的嘴里,会与舌头上的苦味受体结合,让我们尝到苦味。而多酚类物质到了嘴里,则可能与舌头上的蛋白质结合,生成不溶于唾液的沉淀物,然后我们就感觉到了涩。相对来说,绿茶中的儿茶素和咖啡因比较多,所以绿茶比较容易出现苦涩。在红茶里,儿茶素经过氧化和聚合变成茶黄素,能与蛋白质结合的位点变少了,涩味也就降低了。茶黄素与咖啡因的结合在茶黄素自己扎堆之前就会进行。这种结合不仅进一步消耗了茶黄素的结合位点,同时也限制了咖啡因与舌头上苦味受体的结合。于是,与同样固体含量的绿茶茶汤相比,红茶茶汤的苦涩味就往往要低。茶黄素与咖啡因的络合产物溶解度更低,更容易扎堆变大,从而导致冷后浑的出现。因此,许多人认为冷后浑是茶黄素和咖啡因发生反应的结果。在实际的红茶中,咖啡因和茶黄素都存在,所以这样的解释也说得过去。“无事生非”的科学家,会把红茶中的咖啡因去掉,非要看看茶黄素自己能否出现冷后浑——结果是能,只是需要的茶黄素浓度会高一些。冷后浑还有一个名字叫做“茶乳酪”。跟牛奶形成奶酪一样,茶中的茶黄素等成分含量越高,就越容易出现冷后浑。茶黄素是红茶最关键的标志成分——冷后浑意味着它的含量高,“冷后浑是好茶的标志”之说,也就主要是这个原因。在红茶饮料生产中,冷后浑的出现导致产品不均一、外观不合格,风味口感也受到影响。在生产过程中,有一些阶段是以红茶提取物浓缩液的状态存在。因为固体含量高,“茶乳酪”就更容易出现——这会导致有效成分的损失,也为下一步的生产流程带来困难。因此,这个产业需要避免冷后浑的出现——这种需要,也就导致了许多关于冷后浑的研究。科学家们发现,除了咖啡因,茶汤中的钙离子对冷后浑的出现也有显著的作用。他们把茶乳酪拿去分析,发现其中的钙占固体总量的比例,大大高于茶汤中的钙占其固体含量的比例。这是因为,茶汤中的茶黄素带着负电,而钙离子带着正电——类似于卤水点豆腐,钙离子会把本来不想扎堆的茶黄素们拉到一起,让它们沉淀析出。茶中本来就具有不少钙,要避免它导致冷后浑,就需要压制住它的活动。在食品饮料工业中,这可以通过加入“螯合剂”来实现——螯合剂是一些结构特殊的分子,对于钙离子有超级强大的吸引力。只要螯合剂一出现,钙离子们就纷纷投奔而去,茶黄素也就“无钙问津”,不会被它们拉到一起扎堆了。科学家们还发现,如果把糖分子通过“糖苷化”加到茶黄素上,可以增加茶黄素的溶解度、避免冷后浑的出现。红茶制作中的氧化那一步加入一些糖,它们就会在后续的加工过程中结合到茶黄素上去。最后得到的红茶,就不容易出现冷后浑。“冷后浑是优质红茶的标志”是对的,但只是针对正常的红茶。当科学研究让我们对冷后浑有了更深入的认识,就会发现:如果我们不喜欢它,可以通过技术手段避免它的出现;如果我们喜欢它,也可以“捣鬼”促使它的出现。转自:科学松鼠会
各位师兄师姐,有没有拿二级测氨基糖苷类(我做庆大霉素、依替米星和阿米卡星)吗?柱子和流动相用的什么呀?七氟丁酸实验室不给用,HPLC实验室只有紫外和荧光检测器,不能做ELSD和FID。我的课题刚上手,好多不太懂,老师催着摸方法,好纠结。。。谢谢各位师兄师姐~
求具有CNAS资格的维卡校准机构,有资格的情联系: 15228380909 张
【序号】:5【作者】:蔡海瑜郭宝芝刘爱珍 【题名】:宫腔镜下宫腔黏连分离术治疗中重度宫腔粘连疗效的影响因素分析【期刊】:中国实用医刊. 【年、卷、期、起止页码】:2020,47(18)【全文链接】:https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CJFD&dbname=CJFDZHYX&filename=MEDI202018026&uniplatform=NZKPT&v=uWMf5ymM9Gt-fNxIunKklmIbIHQGQIrJn6DySeOk_hakz3ICXj-FKkjRjMTNtKXR
1、雷公藤红素抑制CML细胞增殖作者首先进行了网络药理学分析,以评估在治疗CML方面最有效的天然产物。通过对3882种天然产物进行了网络药理学分析,发现从传统中药“雷公藤”(Tripterygium wilfordii)根皮中提取的五环三萜雷公藤红素在抑制CML方面排名第一。为了验证网络药理学筛选的可靠性,作者在CML细胞中进行细胞活力测定。选择18β-甘草次酸作为阴参,因为它与雷公藤红素的结构最相似,但在3882 种天然产物中预测得分不高,选择17-AAG(HSP90抑制剂,已有文章报道HSP90是雷公藤红素的靶点)和TKI 药物伊马替尼作为阳参。结果表明雷公藤红素、17-AAG和伊马替尼均能有效抑制CML细胞增殖,而18β-甘草次酸几乎不影响细胞生长。作者进一步开展细胞实验,发现雷公藤红素对K562和K562T315I细胞表现出抗增殖活性,诱导细胞凋亡。尽管对雷公藤红素的研究很深入,但尚未系统地鉴定出雷公藤红素在CML中的直接蛋白质靶点,尤其是在耐药性CML细胞中 雷公藤红素抑制CML细胞增殖2、雷公藤红素处理后 K562T315I 细胞的转录组和蛋白质组学分析接着,作者通过RNA 测序发现富集的通路包括铁死亡、蛋白水解调节、响应p53介导的DNA损伤等。作者还进行了蛋白质组学分析雷公藤红素对K562T315I细胞中蛋白质表达水平的调节,下调蛋白主要富集于DNA和RNA代谢途径以及 DNA损伤反应,以及蛋白质加工途径。MCODE分析发现“对DNA损伤刺激的反应”和“对未折叠蛋白的反应”分别是最具特征性的途径。雷公藤红素与其已知靶标HSP90的相互作用可能是“对未折叠蛋白的反应”上调的关键贡献事件。而目前尚未有报道称雷公藤红素的直接蛋白质靶标与“对DNA损伤刺激的反应”途径有关( 雷公藤红素处理后 K562T315I 细胞的定量蛋白质组学分析3、雷公藤红素处理后 K562T315I 细胞的CETSA-MS分析作者接着检测了K562T315I细胞中celastrol处理后可溶性蛋白质水平的变化,在雷公藤红素处理后鉴定了178种差异溶解蛋白质,主要位于DNA中心区域,包括细胞核和线粒体,更具体地说是在DNA损伤位点。此外,“分子伴侣复合物”中溶解度降低,这可能是由于雷公藤红素和HSP90之间的互作所致 雷公藤红素处理后 K562T315I细胞的CETSA-MS分析4、雷公藤红素诱导 K562T315I 细胞DNA损伤对 K562T315I细胞经雷公藤红素处理后总蛋白和可溶性蛋白水平变化的系统分析表明,雷公藤红素主要诱导K562T315I细胞中的DNA损伤和未折叠蛋白反应。因此,作者进行了实验来验证这些观察结果。结果显示雷公藤红素显著诱导γ-H2AX(DNA损伤的常见标志物)的表达,并降低DNA损伤修复相关蛋白FANCD2水平,彗星试验进一步证实了雷公藤红素促进的DNA损伤( 雷公藤红素诱导 K562T315I细胞DNA损伤5、雷公藤红素在K562T315I细胞中的靶点鉴定然后,作者在细胞裂解物中开展质谱耦合等温剂量反应-细胞热位移分析(MS-ITDR-CETSA)实验,以确定雷公藤红素的直接蛋白质靶标,特别是那些参与DNA损伤反应的蛋白质靶标。在检测到的3393种蛋白质中,有12种蛋白质表现出热稳定性的显著变化,代表了最有潜力且可信度高的靶标蛋白质。值得注意的是,雷公藤红素的已知靶标HSP90 (HSP90AA1和HSP90AB1) 的热稳定性仅表现出很小的变化,并且没有超过阈值。对这12个潜在靶标和定量蛋白质组学以及CETSA-MS分析的差异蛋白进行PPI分析,发现 YY1均为最紧密相关的蛋白质。因此,YY1与所有这些DEP/DSP的关联节点数量最多,并且可能是与DNA损伤相关的最重要的靶标。现有研究表明,YY1作为转录因子,可以调节参与DNA修复和细胞存活的各种蛋白质的表达,以响应DNA损伤。此外,HMCES已被确定为通过屏蔽脱碱基位点来保护基因组完整性免受氧化碱基损伤的关键蛋白。因此,作者继续通过蛋白质印迹结合细胞热位移分析(WB-CETSA)验证了celastrol与YY1和HMCES的互作。同样,报道的阳性对照HSP90蛋白也显示出明显的热稳定性增加( 雷公藤红素在K562T315I细胞中的靶点鉴定6、雷公藤红素与YY1和HMCES相互作用的验证为了进一步验证celastrol与YY1和HMCES的直接相互作用,合成了可点击炔烃标签功能化celastrol探针(Cel-P),该探针保留了celastrol对K562T315I细胞的抑制活性。利用该探针开展Pulldown实验发现Cel-P 能够成功地从细胞中拉下HMCES和HSP90蛋白,但由于尚不清楚的原因,在蛋白质印迹膜上的下拉样本中未检测到YY1。随后,表达并纯化重组YY1(rYY1)蛋白,发现随着Cel-P浓度的增加,rYY1的标记以剂量依赖性方式增加 雷公藤红素与YY1和HMCES相互作用的验证7、雷公藤红素通过靶向YY1和HMCES诱导DNA损伤在验证了celastrol与YY1和HMCES之间的相互作用后,作者继续在K562T315I细胞中敲低 YY1或HMCES。结果显示YY1或HMCES的敲低显著增加了DNA损伤的发生率,同时影响细胞生长,增强细胞对celastrol的敏感性。此外,与HMCES相比,YY1敲低对细胞的影响更为显著,表明YY1发挥着更为重要的作用。对接分析显示,与HMCES相比,celastrol对YY1的亲和力略强,且Celastrol与YY1上的Leu132和Val316形成氢键,与HMCES的Glu127、Arg130和Arg137形成氢键 雷公藤红素通过靶向 YY1 和 HMCES 诱导 DNA 损伤鉴于YY1在雷公藤红素诱导的DNA损伤反应中发挥关键作用,作者对YY1蛋白进行了进一步实验。发现YY1过表达对细胞生长没有显著影响,但减轻了雷公藤红素引起的细胞死亡和DNA损伤,且通过裂解的PARP1和Caspase-3水平发现YY1表达与雷公藤红素诱导的细胞凋亡呈负相关。使用双荧光素酶报告基因发现雷公藤红素显著抑制了YY1的转录活性,BLI结合试验发现celastrol 可以与 rYY1 结合(图8)。图8 YY1在雷公藤红素诱导的 K562T315I细胞DNA损伤和细胞死亡中起关键作用总结研究使用多组学方法对雷公藤红素的作用机理进行了系统研究,利用蛋白质组范围的无标记靶标反卷积方法MS-CETSA来识别雷公藤红素的蛋白质靶标。研究不仅验证了雷公藤红素通过靶向HSP90来诱导未折叠蛋白反应,而且还发现它通过直接靶向耐药 K562T315ICML 细胞中的YY1和HMCES来诱导DNA损伤(图9)。研究有助于更好地理解雷公藤红素的多方面机制。研究提供了一种有效的系统药理学工作流程范例,该范例集成了网络药理学分析、蛋白质丰度和溶解度测量以及 MS-CETSA,以揭示任何天然产物或活性化合物的作用机理。
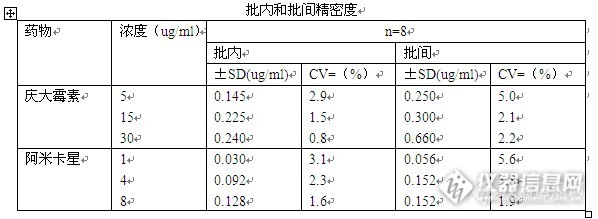
高效液相色谱法快速测定血清中的庆大霉素和阿米卡星庆大霉素和阿米卡星是用于严重革兰氏阴性细菌感染治疗的氨基糖苷类抗生素。像其他氨基糖苷类抗生素一样,庆大霉素和阿米卡星的有效治疗范围狭窄容易导致肾脏毒性和耳毒性。因此,监测庆大霉素和阿米卡星血清中的水平对于安全和有效的临床应用十分重要。最近,高压液相色谱法已被报道用于氨基糖苷类抗生素的测定。多数测定需要柱前或柱后衍生荧光检测。这些方法涉及耗时的预处理,如血清中氨基糖苷类抗生素的柱萃取以及溶剂。本实验开发了一种较为简单的预处理方法来测定血清中的庆大霉素和阿米卡星,并与荧光偏振免疫测定法进行了比较。材料和方法:庆大霉素和阿米卡星购自药店,邻苯二甲醛(瑞尔丰化工),2-巯基乙醇、庚烷磺酸钠、1,2-乙烷二磺酸二钠、色谱乙腈、蒸馏水。蛋白质沉淀剂(10mM辛烷磺酸钠溶解于3.5%高氯酸中)。庆大霉素的流动相为含有22 mM的1,2 - 乙烷二磺酸二钠和5mM辛烷磺酸钠的水 - 乙腈混合物(80:20,体积/体积),用乙酸调节pH至约3.5。阿米卡星的流动相为含有37 mM的1,2 - 乙烷二磺酸二钠和5mM辛烷磺酸钠的水 - 乙腈混合物(80:20,体积/体积),用乙酸调节pH至约3.5。仪器和色谱条件:安捷伦高效液相色谱仪1200,UV检测器,色谱柱:安捷伦Agilent液相色谱柱4.6﹡150﹡5u,流动相的流速保持在1毫升/分钟。邻苯二甲醛试剂用泵以0.6毫升/分钟的流速。实验部分:将50ul血浆加入到50ul蛋白质沉淀剂,涡旋混合数秒后,使用KM-15200离心机离心2分钟(15000rpm),离心后上清液进样。正常血清,分别加入已知量的庆大霉素和阿米卡星,进行分析,根据加入量和峰面积绘制标准曲线。荧光偏振免疫通过市售的试剂盒进行。结果:图1为庆大霉素的标准色谱图,庆大霉素加入对照血清的色谱图以及对照血清的色谱图。(其中交标庆大霉素的含量为15ug/ml)http://ng1.17img.cn/bbsfiles/images/2014/09/201409300949_516466_2188679_3.jpg图2为阿米卡星的标准色谱图,阿米卡星加入对照血清的色谱图以及对照血清的色谱图,经过蛋白质沉淀剂处理后的血清和未处理的色谱图基本形同,皆未见到影响抗生素检测的干扰杂质峰。http://ng1.17img.cn/bbsfiles/images/2014/09/201409300949_516467_2188679_3.jpg以峰面积作为纵坐标,血清中的药物浓度为横坐标得到庆大霉素的线性标准曲线(3-50ug/ml)为y =0.979x - 0.006,阿米卡星的线性标准曲线(0.5-10ug/ml)为y= 0.998x - 0.04根据庆大霉素和阿米卡星在血清中不同浓度的重复测定,得到庆大霉素和阿米卡星的批内变异系数分别为0.8 -2.9%和1.6-3.1%,批间变异系数为2.0-5.0%及1.9-5.6%。http://ng1.17img.cn/bbsfiles/images/2014/09/201409300949_516468_2188679_3.jpg回收率试验:分别将15ug/ml的庆大霉素水溶液加入含有15ug/ml阿米卡星的血清中和将4ug/ml的阿米卡星水溶液加入4ug/ml的血清中。将它们的峰面积进行比较。基于三个样品的平均值,庆大霉素和阿米卡星的分别为99.0%和98.5%.通过该方法获得的结果与用荧光偏振免疫测定法进行了比较,回归方程和相关系数分别为庆大霉素:y=1.027x - 1.090,n =44和r=0.996。阿米卡星y=0.998x - 0.206, n =42和r=0.957。讨论:本实验采用了蛋白质沉淀剂,排除干扰物,再将样品进行分析,优化的分析方法与荧光偏振免疫测定法进行了比较种,该方法方法的优点是速度快,操作简便,重复性好。因此,该方法可用于该药物的分析。
影响卡尔-费休(Karl-Fisher)滴定法测水精度的几个因素方 建 安(中科院南京土壤研究所 南京 210008)一.引 言 1935年卡尔-费休(Karl-Fisher)首先提出了一种利用容量分析测定水的方法,即通常的卡尔-费休法, 它是利用碘氧化二氧化硫时需要定量的水的原理测定液体、固体和气体样法中的含水量。被许多国家定为标准分析方法, 用来校正其它分析方法和测量仪器。 因此用Karl-Fisher法测定水份含量对控制生产过程和产品质量有很好的效果。我们研制和生产的《FJA-1型常规分析仪器工作站》中的微机控制的卡尔-费休自动滴定仪的软件是根据国家标准GB/T13753-92编制而成的,它具有测定精度好,软件功能多,显示、打印和储存测定结果与曲线, 分析者可修改各种参数等特点。但测定的结果正确与否是由多种因素决定的, 除了有一个好的测定仪器外,同时考虑其它各方面的因素,才能得到可靠、正确的数据。现综合有关文献和作者的经验,把有关问题叙述如下,供有关分析者参考。二.有关问题 (一).应用范围 Karl-Fisher滴定法可适用于多种有机和无机物中含水的测定。由于各种化合物性质的差异, 可分为能直接进行测定和不能直接进行测定两类。可以直接测定的主要有机和无机化合物如表1所示。 表1 无干扰的有机和无机化合物 化合物种类 举 例 1.无机化合物(1).有机酸 Na(CH3 )SO4 ,Ba(OOCCH3 )2 ,K2 C2 O4 ,VO2 (OOCCH3 )2 ,Na2 C2 H4 O6 (2).无机酸 NH4 PO4 ,CaCl2 ,NaHSO4 ,Na2 SO4 ,KF,NH4 NO3 ,MgSO4 , Na2 SO4 ,KSCN,FeSO4 ,Al2 (SO4 )3 KSO4 ,CaHPO4 , NaI,CaCO3 ,FeF3 ,VO2 (NO3 )2 (3).酸式氧化物 SiO2 ,Al2 O3 (4).无机酸和酸酐 SO2 ,HI,HF,HNO3 ,HCN,H2 SO4 ,HSO3 ,NH2 2.有机化合物(1).酸 羧酸,羧基酸,氨基酸,磺酸(2).醇 一元醇,多元醇,酚(3).酯 酸酯,正酸酯,氨基甲酸酯内酯,无机酸酯(4).稳定的羟基化合物 糖,甲醛,二苯基乙二酮,二苯乙醇酮,二氯乙醛(5).缩醛,醚 缩甲醛,二乙醚(6).烃 饱和与不饱合脂族和芳香族化合物(7).酸酐和酰卤 乙酸酐,苯甲酰氯(8).卤化物 卤代烷(9).过氧化合物 过氧化氢,二烷基过氧化物(10).含氮化合物 胺, 胺,腈(11) .含硫化合物 硫化物,硫氰酸盐,硫醚,磺原酸盐,二硫化氨基甲酸脂不能直接测定的主要有机和无机化合物如表2所示。 表2 有干扰的有机和无机化合物 化合物种类 干 扰 性 质 1.无机化合物(1).金属氢氧化物及氧化物 与费休试剂定量反应(2).碳酸盐及酸式碳酸盐 同上(3).醋酸铅,碱式氨 反应不完全(4).硼酸及氧化物 与碘反应(5).铬酸及重铬酸 非定量反应(6).钴氨络合物 同上(7).铜的氯化物及硫酸盐 被HI定量还原(8).氯化铁 与费休试剂定量反应(9).硫化氢及硫化钠 反应不确定(10).羟胺 与费休试剂部分反应(11).磷钼酸 反应不完全(12).甲基硅烷醇(R3 SiOH) 与费休试剂定量反应(13).硫代硫酸盐 同上(14).二氯化锡 同上(15).二氯化氧锆 反应不完全 2.有机化合物(1).活泼羰基化合物 形成缩醛(2).过氧化合物 与试剂中的SO2 反应(3).抗坏血酸 被碘定量氧化(4).硫醇 同上(5).醌 被HI定量还原(6).二酰基过氧化物 被HI还原(7).Dimethylo Lnred 凝聚 从上述表格中可以得出以下几点意见:1. 卡尔费休测水法适用于许多无机化合物和有机化合物中含水量的测定。 2.由于化合物性质的差异, 可分为能直接进行测定和不能直接进行测定两类。因此要求分析工作者在测定某种化合物中的水时, 首先考虑它属于那一类,如果是后者,而又采用直接测定,则将产生很大的测定误差或根本无法进行测定。 3.如果要对不能进行直接测定的化合物中的水进行测定时, 必须采用合适的方法消除各种干扰因素,达到正确测定的目的。(二).仪器的标定物质卡尔费休滴定仪通常用甲醇-水标准溶液,含水酒石钠, 蒸馏水,含饱和水甲苯等类物质作为标准对方法的可靠性进行校验。含水酒石酸钠是一种常用的含水标准物质,理论含水量为15.66%,在105℃加热失重为15.65±0.02%,长期暴露于湿度为20~70%的空气中,增重为0.01~0.09%。 用含饱和水的甲苯和纯水的标定结果也是满意的。当然,最简单还是用甲醇-水标准溶液。(三).取样与取样量 在做分析取样时应尽量取混合均匀后的代表性样品,并应观察容器底部游离水分存在的情况。在用注射器抽取试样时,抽取速度不能太快,否则有可能空气进入注射器形成气泡, 造成进样误差。在分析前如果发现试样与容器有乳浊现象,或瓶壁有微小水珠析出时,则必须用乙二醇抽提法进行分析。具体方法如下: 将预先干燥的细口瓶中加入三分之一试样加盖密闭, 在工业分析天平上称准至0.1克,然后称入2至3倍于重量的乙二醇用力摇动15分种,静止分层后,用注射器通过试样层吸取0,25~1.0mL乙二醇, 测定其含水量,同时也测定乙二醇的原始水含量。分析完毕后将瓶中试样倒掉,洗净烘干,在天平上称准至0.1克,根据上述三次称量之差,求出试样和乙二醇的重量,就可求出试样的含水量。 在进样前首先用侍分析试液清洗注射器5~7次,然后根据试样含水量的多少决定取样量大小,通常按表3规定的注射器取样量抽取<0.1~5mL试样。 表3 取样量参考数据 试样含水量(ppm) 取样量(mL) 0-10 2-5 10-100 1-2 100-1000 0.1-1 >1000 <0.1 从表3中可以看到含水量大的物质取样量小,反之取样量要大,否则将产生较大的测量误差。同时特别要注意进样时注射器中是否存在小气泡, 以防产生严重的测量误差。(四).测定精度 卡尔费休滴定法测定物质含水量范围很宽从几个ppm到100%, 对精度的要求是根据含水量大小决定的。通常要求平行测定两个结果与算术平均的差数不应大于下列数值: 含水量(ppm) 允许差值 1-10 1ppm 10-50 算术平均值±10% >50 算术平均值±5% 在进行分析时,取两次测定结果的算术平均值作为分析结果。(五).影响测定精度的几个原因 除了上述测定样品的性质,测定的方法,标定物质的选用,取样方法和进样量的大小影响测定精度外,还必须注意以下几个问题,才能保证测定精度。1. 由于卡尔费休滴定试剂很容易吸收水分,因此要求滴定剂发送系统的滴定管和滴定池(测量池)等采取较好的密封系统。否则由于吸湿现象造成终点长时间的不稳定和严重的误差。2. 卡尔费休试剂的滴定度的大小, 根据试液含水量的多少来决定。在测定含水量较大的试液时,卡尔费休试剂的滴定度应该选得大一些,这样在保证测定精度(<5%)的前提下,可以加快测定速度。但在测定试液含水量较小时,卡尔费休试剂的滴定度就应该选得小一些和滴定管的最小读数小一些, 否则将产生较大的测定误差。如果滴定管的最小读数为0.01mL,卡尔费休试剂的滴定度为2.5mg/mL,则试剂一滴误差将产生0.025mg(25ppm)的测量误差。如果试剂的滴定度1.00mg/mL,则试剂一点误差将产生0.015mg(15ppm)的测量误差。 3.卡尔费休滴定法测定水的终点判别方有: (1).依靠人的视觉观察溶液颜色突变的目视法 (2).依靠观察电流表偏转突变至一定值并稳定一段时间如60秒作为滴定终点的永停终点法(硬件滴定) (3).以永停终点法又称为死停终点法(dead stop end-point method)为基础,微机自动控制的软件滴定三种方法。 目视终点法是指示终点最简单一种方法,可以省去滴定仪中的指示系统装置,在常量滴定中可以获得比较满意的测定结果,但在毫克当量以下物质的测定中,这种方法的灵敏度和准确度比较差, 一般都采用比较灵敏的电化学方法。第二种与第三种方法都是电化学方法,它有快速、灵敏而且准确度又比较高,易实现自动化等优点,通常可测定各类样品中几个ppm到百分之几十的水分。 4.滴定试剂的发送头的结构与位置也是滴定误差的一个非常重要的因素。通常要求发送滴定头内径和滴定头要做得很细,目的防止滴定剂的挂滴现象,保证测量精度。在滴定头插入样品溶液中时, 滴定头的液界处有可能发生化学反应而影响测定精度。 5.在滴定时搅拌要均充分且均匀。在滴定粘度较大的样品溶液时更要注意搅拌的充分和一致, 包括磁力搅拌器的速度要一致和滴定池中的液面高度大体相同,这样才能得到较好的测定精度。 6.在进样时,要防止注射器头受外界的污染而影响测定结果,如操作者呼气和擦注射器头时的污染等。同时要防止进样
[url=http://www.ldteq.com/article/3080.html]Glenair[/url][font=宋体][font=宋体]超薄型[/font][font=Calibri]MouseBud[/font][font=宋体]?连接器重量较轻,具备屏蔽功能性,致力于在极端环境下使用需求设计。卡扣锁定、扳机释放连接器插座具备弹簧镀金接触点、不锈钢板外壳自驱动接口耦合环。电缆安装版本根据充分组装、屏蔽、包括成型并通过[/font][font=Calibri]100%[/font][font=宋体]检测。卡扣锁定、扳机释放连接器受到全面保护,不受粉尘、潮湿渗入、电磁干扰及多种其它环境效应的影响。弹簧加载接触系统的额定插拔次数为[/font][font=Calibri]2000[/font][font=宋体]次。[/font][font=Calibri]860[/font][font=宋体]系列连接器主要用于高速数据、开关电源、视频和音频。[/font][/font][font=宋体][font=宋体]具有[/font][font=宋体]“热靴”型触点的支架安装[/font][font=Calibri]MouseBud[/font][font=宋体]插座主要用于极端环境,包括军队可穿戴电子设备和战术装备。接触点易于清洗且不易损坏。卡扣锁定、扳机释放连接器满足[/font][font=Calibri]MIL-STD-810G[/font][font=宋体]需求,能够在极端环境中(不管是插接或是未插接)实现安全可靠的性能。卡扣锁定、扳机释放连接器根据不锈钢板锁紧螺母连接至面板。氟硅橡胶[/font][font=Calibri]O[/font][font=宋体]形圈提供面板密封性。选用[/font][font=Calibri]PC[/font][font=宋体]尾端子端接至柔性电路板,或选择焊杯端子实现电缆连接。[/font][/font][font=宋体][font=Calibri]MouseBud[/font][font=宋体]直角插座具备单件式“眼镜蛇式”连接器外壳紧密连接的盖子。外壳盖子之间带有橡胶垫圈,以避免潮湿渗入。与后面板安装插座搭配时的总高度远远低于[/font][font=Calibri]1/2[/font][font=宋体]英寸。[/font][/font][font=宋体][font=宋体]包括成型[/font][font=Calibri]MouseBud[/font][font=宋体]线组主要有两种标准版本。[/font][font=Calibri]1[/font][font=宋体]型线组选用热塑性聚氨酯绝缘套管和聚酰胺包括成型。具有热塑性橡胶[/font][font=Calibri](TPV)[/font][font=宋体]绝缘套管和包括成型的[/font][font=Calibri]2[/font][font=宋体]型线组能够在低至[/font][font=Calibri]-55[/font][font=宋体]°[/font][font=Calibri]C[/font][font=宋体]温度下提供优异的冷弯性能。[/font][/font][font=Calibri]Glenair[/font][font=宋体]连接器是全球知名的高端品牌,广泛应用于航天航空、军用、舰载、精密制造等场合。深圳市立维创展科技有限公司,授权代理销售[/font][font=Calibri]Glenair[/font][font=宋体]产品,并提供技术支持。欢迎咨询。[/font][font=宋体]详情了解[/font][font=Calibri]Glenair[/font][font=宋体]请点击:[/font][url=http://www.ldteq.com/brand/72.html][font=Calibri]http://www.ldteq.com/brand/77.html[/font][/url][font=Calibri] [/font][table][tr][td][font=Calibri]860-006P[/font][/td][td][font=Calibri]860-007P[/font][/td][td][font=Calibri]860-004R[/font][/td][td][font=Calibri]860-005R[/font][/td][/tr][/table][font=Calibri] [/font]
影响卡尔-费休(Karl-Fisher)滴定法测水精度的几个因素一.引 言1935年卡尔-费休(Karl-Fisher)首先提出了一种利用容量分析测定水的方法,即通常的卡尔-费休法, 它是利用碘氧化二氧化硫时需要定量的水的原理测定液体、固体和气体样法中的含水量。被许多国家定为标准分析方法, 用来校正其它分析方法和测量仪器。 因此用Karl-Fisher法测定水份含量对控制生产过程和产品质量有很好的效果。我们研制和生产的《FJA-1型常规分析仪器工作站》中的微机控制的卡尔-费休自动滴定仪的软件是根据国家标准GB/T13753-92编制而成的,它具有测定精度好,软件功能多,显示、打印和储存测定结果与曲线, 分析者可修改各种参数等特点。但测定的结果正确与否是由多种因素决定的, 除了有一个好的测定仪器外,同时考虑其它各方面的因素,才能得到可靠、正确的数据。现综合有关文献和作者的经验,把有关问题叙述如下,供有关分析者参考。二.有关问题(一).应用范围Karl-Fisher滴定法可适用于多种有机和无机物中含水的测定。由于各种化合物性质的差异, 可分为能直接进行测定和不能直接进行测定两类。可以直接测定的主要有机和无机化合物如表1所示。表1 无干扰的有机和无机化合物化合物种类 举 例1.无机化合物(1).有机酸 Na(CH3 )SO4 ,Ba(OOCCH3 )2 ,K2 C2 O4 ,VO2 (OOCCH3 )2 ,Na2 C2 H4 O6 (2).无机酸 NH4 PO4 ,CaCl2 ,NaHSO4 ,Na2 SO4 ,KF,NH4 NO3 ,MgSO4 , Na2 SO4 ,KSCN,FeSO4 ,Al2 (SO4 )3 KSO4 ,CaHPO4 , NaI,CaCO3 ,FeF3 ,VO2 (NO3 )2 (3).酸式氧化物 SiO2 ,Al2 O3 (4).无机酸和酸酐 SO2 ,HI,HF,HNO3 ,HCN,H2 SO4 ,HSO3 ,NH2 2.有机化合物(1).酸 羧酸,羧基酸,氨基酸,磺酸(2).醇 一元醇,多元醇,酚(3).酯 酸酯,正酸酯,氨基甲酸酯内酯,无机酸酯(4).稳定的羟基化合物 糖,甲醛,二苯基乙二酮,二苯乙醇酮,二氯乙醛(5).缩醛,醚 缩甲醛,二乙醚(6).烃 饱和与不饱合脂族和芳香族化合物(7).酸酐和酰卤 乙酸酐,苯甲酰氯(8).卤化物 卤代烷(9).过氧化合物 过氧化氢,二烷基过氧化物(10).含氮化合物 胺, 胺,腈(11) .含硫化合物 硫化物,硫氰酸盐,硫醚,磺原酸盐,二硫化氨基甲酸脂不能直接测定的主要有机和无机化合物如表2所示。表2 有干扰的有机和无机化合物化合物种类 干 扰 性 质1.无机化合物(1).金属氢氧化物及氧化物 与费休试剂定量反应(2).碳酸盐及酸式碳酸盐 同上(3).醋酸铅,碱式氨 反应不完全(4).硼酸及氧化物 与碘反应(5).铬酸及重铬酸 非定量反应(6).钴氨络合物 同上(7).铜的氯化物及硫酸盐 被HI定量还原(8).氯化铁 与费休试剂定量反应(9).硫化氢及硫化钠 反应不确定(10).羟胺 与费休试剂部分反应(11).磷钼酸 反应不完全(12).甲基硅烷醇(R3 SiOH) 与费休试剂定量反应(13).硫代硫酸盐 同上(14).二氯化锡 同上(15).二氯化氧锆 反应不完全2.有机化合物(1).活泼羰基化合物 形成缩醛(2).过氧化合物 与试剂中的SO2 反应(3).抗坏血酸 被碘定量氧化(4).硫醇 同上(5).醌 被HI定量还原(6).二酰基过氧化物 被HI还原(7).Dimethylo Lnred 凝聚从上述表格中可以得出以下几点意见:1. 卡尔费休测水法适用于许多无机化合物和有机化合物中含水量的测定。2.由于化合物性质的差异, 可分为能直接进行测定和不能直接进行测定两类。因此要求分析工作者在测定某种化合物中的水时, 首先考虑它属于那一类,如果是后者,而又采用直接测定,则将产生很大的测定误差或根本无法进行测定。3.如果要对不能进行直接测定的化合物中的水进行测定时, 必须采用合适的方法消除各种干扰因素,达到正确测定的目的。(二).仪器的标定物质卡尔费休滴定仪通常用甲醇-水标准溶液,含水酒石钠, 蒸馏水,含饱和水甲苯等类物质作为标准对方法的可靠性进行校验。含水酒石酸钠是一种常用的含水标准物质,理论含水量为15.66%,在105℃加热失重为15.65±0.02%,长期暴露于湿度为20~70%的空气中,增重为0.01~0.09%。 用含饱和水的甲苯和纯水的标定结果也是满意的。当然,最简单还是用甲醇-水标准溶液。(三).取样与取样量在做分析取样时应尽量取混合均匀后的代表性样品,并应观察容器底部游离水分存在的情况。在用注射器抽取试样时,抽取速度不能太快,否则有可能空气进入注射器形成气泡, 造成进样误差。在分析前如果发现试样与容器有乳浊现象,或瓶壁有微小水珠析出时,则必须用乙二醇抽提法进行分析。具体方法如下:将预先干燥的细口瓶中加入三分之一试样加盖密闭, 在工业分析天平上称准至0.1克,然后称入2至3倍于重量的乙二醇用力摇动15分种,静止分层后,用注射器通过试样层吸取0,25~1.0mL乙二醇, 测定其含水量,同时也测定乙二醇的原始水含量。分析完毕后将瓶中试样倒掉,洗净烘干,在天平上称准至0.1克,根据上述三次称量之差,求出试样和乙二醇的重量,就可求出试样的含水量。在进样前首先用侍分析试液清洗注射器5~7次,然后根据试样含水量的多少决定取样量大小,通常按表3规定的注射器取样量抽取<0.1~5mL试样。表3 取样量参考数据试样含水量(ppm) 取样量(mL)0-10 2-510-100 1-2100-1000 0.1-1>1000 <0.1 从表3中可以看到含水量大的物质取样量小,反之取样量要大,否则将产生较大的测量误差。同时特别要注意进样时注射器中是否存在小气泡, 以防产生严重的测量误差。(四).测定精度卡尔费休滴定法测定物质含水量范围很宽从几个ppm到100%, 对精度的要求是根据含水量大小决定的。通常要求平行测定两个结果与算术平均的差数不应大于下列数值:含水量(ppm) 允许差值1-10 ?1ppm10-50 算术平均值±10%>50 算术平均值±5% 在进行分析时,取两次测定结果的算术平均值作为分析结果。(五).影响测定精度的几个原因除了上述测定样品的性质,测定的方法,标定物质的选用,取样方法和进样量的大小影响测定精度外,还必须注意以下几个问题,才能保证测定精度。1. 由于卡尔费休滴定试剂很容易吸收水分,因此要求滴定剂发送系统的滴定管和滴定池(测量池)等采取较好的密封系统。否则由于吸湿现象造成终点长时间的不稳定和严重的误差。2. 卡尔费休试剂的滴定度的大小, 根据试液含水量的多少来决定。在测定含水量较大的试液时,卡尔费休试剂的滴定度应该选得大一些,这样在保证测定精度(<5%)的前提下,可以加快测定速度。但在测定试液含水量较小时,卡尔费休试剂的滴定度就应该选得小一些和滴定管的最小读数小一些, 否则将产生较大的测定误差。如果滴定管的最小读数为0.01mL,卡尔费休试剂的滴定度为2.5mg/mL,则试剂一滴误差将产生0.025mg(25ppm)的测量误差。如果试剂的滴定度1.00mg/mL,则试剂一点误差将产生0.015mg(15ppm)的测量误差。3.卡尔费休滴定法测定水的终点判别方有: (1).依靠人的视觉观察溶液颜色突变的目视法 (2).依靠观察电流表偏转突变至一定值并稳定一段时间如60秒作为滴定终点的永停终点法(硬件滴定) (3).以永停终点法又称为死停终点法(dead stop end-point method)为基础,微机自动控制的软件滴定三种方法。目视终点法是指示终点最简单一种方法,可以省去滴定仪中的指示系统装置,在常量滴定中可以获得比较满意的测定结果,但在毫克当量以下物质的测定中,这种方法的灵敏度和准确度比较差, 一般都采用比较灵敏的电化学方法。第二种与第三种方法都是电化学方法,它有快速、灵敏而且准确度又比较高,易实现自动化等优点,通常可测定各类样品中几个ppm到百分之几十的水分。4.滴定试剂的发送头的结构与位置也是滴定误差的一个非常重要的因素。通常要求发送滴定头内径和滴定头要做得很细,目的防止滴定剂的挂滴现象,保证测量精度。在滴定头插入样品溶液中时, 滴定头的液界处有可能发生化学反应而影响测定精度。5.在滴定时搅拌要均充分且均匀。在滴定粘度较大的样品溶液时更要注意搅拌的充分和一致, 包括磁力搅拌器的速度要一致和滴定池中的液面高度大体相同,这样才能得到较好的测定精度。6.在进样时,要防止注射器头受外界的污染而影响测定结果,如操作者呼气和擦注射器头时的污染等。同时要防止进样
按照国标方法测定尿素中缩二脲,缩二脲在硫酸铜、酒石酸钾钠的碱性溶液中应该生成紫红色配合物,在波长为550 nm处测定其吸光度。可是我们反复做 反应得不到紫红色的络合物,是蓝色的配合物,请教做个这个实验的人,帮忙分析下原因/国标上传在这里了(zhouyuhu)
咖啡产业在全球经济中扮演着主要的角色,其对于环境的影响也至关重要,每年全球会生产超过20亿吨的咖啡副产品;当咖啡豆焙烤干燥后,咖啡谷皮(即咖啡豆的表皮)通常在加工过程中会被移去, 同时咖啡渣也会被直接丢弃。传统观念认为这些副产品:咖啡渣和咖啡谷皮并不会存在太多的实用价值及应用,而咖啡渣有时候还可以被自制成为皮肤去角质的甘醇酸(Exfoliants)或者作为清洁产品。近日,刊登在国际杂志LWT-Food Science and Technology上的一篇研究论文中,来自格拉纳达大学的科学家们开始进行研究来确定哪些咖啡副产品可以被回收利用作为营养物质,从而帮助减少咖啡副产品的浪费量;文章中,研究者阐明咖啡渣和咖啡谷皮具有强大的抗氧化和抗菌特性,因为其富含纤维和酚类,的确该研究发现也揭示了咖啡渣的抗氧化效力是维生素C的500倍以上,因此利用其作为功能性食品将带来巨大的健康效益。研究者Rufian Henares教授说道,咖啡渣等副产品中还包括高水平的蛋白黑素,其在焙烧过程中常常会产生同时使得咖啡呈现咖啡色,这些蛋白黑素的生化特性或许可以进行一系列实际的应用,比如预防有害的病原体在食品中生长;然而如果我们可以利用咖啡副产品的有益效应,那么首先我们需要移去这种蛋白黑素,因为其会干预副产品的有益作用。最后研究者总结道,对咖啡副产品的处理或可使其作为新型的食品成分进行潜在地再循环,这或许可以明显较少丢弃的咖啡副产品带来的环境效应。来自经济学界和金融学界的研究者近日呼吁进行一项计划来加速对咖啡副产品潜在价值的评估,以便其可以更好地被利用。

[b][color=#cc0000]一起欣赏意大利托斯卡纳的风光15[/color][color=#cc0000] 意大利中部的托斯卡纳,文艺复兴的发祥地,先后涌现了以乔托、米开朗基罗、达芬奇、拉斐尔为代表的艺术家。[/color][/b][img=,690,635]https://ng1.17img.cn/bbsfiles/images/2021/12/202112162126518341_2757_1841897_3.jpg!w690x635.jpg[/img]
测定苯巴比妥、苯妥英钠和卡马西平







 我要推广仪器
我要推广仪器
 下载APP
下载APP