我想取1ml氢氟酸。请教大虾如何取
因为氢氟酸有溶解玻璃的特性,普通的玻璃量器都不能量取氢氟酸,做氢氟酸试验时大家是如何量取和转移氢氟酸的?
在向聚四氟乙烯坩埚内加1毫升氢氟酸、或1毫升硝酸时,大家用什么工具定量取浓酸呢!弱弱的问下啊!!http://simg.instrument.com.cn/bbs/images/default/em09502.gif
在测定土壤样品中稀土元素时如果使用了氢氟酸是不是会导致结果偏低?为什么?
求助:用氢氧化钠中和后的乙酸钙为何呈现黄色?在测定土中阳离子交换量时,配制好0.5mol/l的乙酸钙是无色,移取50ml,加入酞指示剂,用0.02mol/l氢氧化钠滴定至微红色(极浅的红色),计算出应该加入到乙酸钙中2mol/l氢氧化钠的量,但经常是还没有加够氢氧化钠的时候,就出现了黄色。加完后是比较深的黄色,显然这个颜色极不正常。这种状况已经出现多次。想问一下大家,这种情况是乙酸钙的质量问题吗?你们又是用的哪个厂家的乙酸钙呢?
近日测陶瓷制品的 砷样品用 4%乙酸浸泡,室温,24h取浸泡液 20mL,加入 (1+1)HCl 5mL,5%硫脲-5%Vc 10mL用水定容至 50mL仪器:海光 AFS-230E(手动进样)载流 5%HCl,还原剂 2%KBH4 + 0.5%NaOH载气400, 屏蔽气1000,断续流动程序使用仪器默认值但是在样品进入反应环之后,产生大量泡沫,废液来不及排走,泡沫竟然充满一级气液分离器之后,连二级气液分离器(水封)也充满了,为了避免水汽冲上原子化器,只好不停用针筒从水封中抽掉废液……汗啊-_-!!这是我第一次做含有乙酸的样品,泡沫超多,而且产生得很快,来不及排走。而以前做过的样品中只有盐酸,却不会产生泡沫,废液都能及时排走。请问哪位前辈 可以解释一下,乙酸对生成氢化物反应的过程会产生哪些影响??怎样才可以减少泡沫产生?
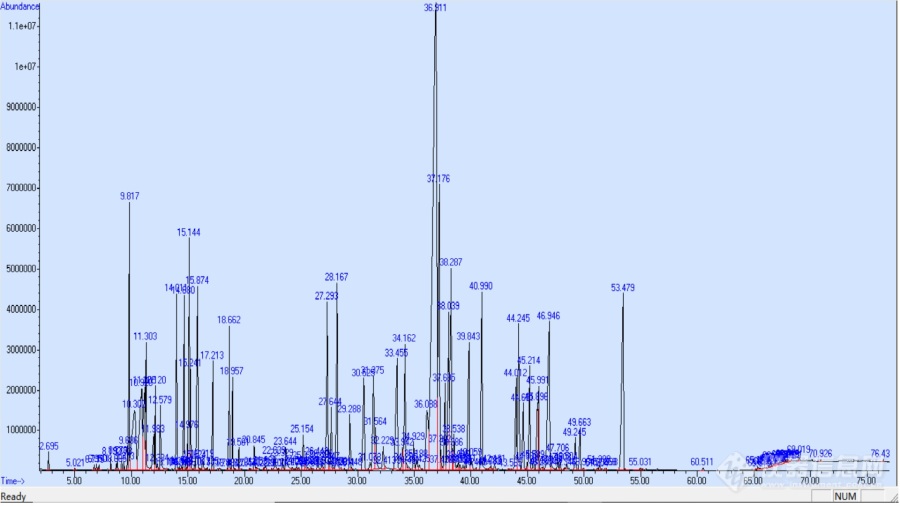
[align=center]未分离峰定量示例2----二氢茉莉酮酸甲酯和龙涎酮合峰处理[/align]网友发帖:日化香精中的龙涎酮和二氢茉莉酮酸甲酯如何更好地定量,参考链接[url]https://bbs.instrument.com.cn/topic/7403742[/url]问题是:请教各位老师,在日化香精中,龙涎酮与二茉峰重合,特征离子也重合,大家是如何很好地定量的?几个网友也参与提出好的建议,有提取离子定量,有amdis等处理方式。下面讨论一下利用单一离子面积计算估计化合物面积的方法来进行定量。[b]1. 样品合峰初步分析[/b]此样品总离子图如下:[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/11/201911301117086589_9865_1615838_3.jpg!w690x387.jpg[/img][align=center]图1 总[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]图TIC[/align]*********************************************************************其中36.91min为二氢茉莉酮酸甲酯和龙涎酮合峰[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/11/201911301117514992_6733_1615838_3.jpg!w690x387.jpg[/img][align=center]图2 36.91min二氢茉莉酮酸甲酯和龙涎酮合峰色谱图[/align]***********************************************************************36.91分钟色谱图的质谱图如下:[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/11/201911301118475898_9906_1615838_3.jpg!w690x387.jpg[/img][align=center]图3 36.91分钟色谱图的质谱图[/align]****************************************************************************经过检索36.91分钟为二氢茉莉酮酸甲酯和龙涎酮的离子加和:[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/11/201911301119357832_3612_1615838_3.jpg!w690x387.jpg[/img][align=center]图4 36.91分钟检索结果[/align]****************************************************************************提取两个化合物的最大离子m/z191和m/z83得到两个化合物的分布图如下:[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/11/201911301121070458_9969_1615838_3.jpg!w690x387.jpg[/img][align=center]图5 提取两个化合物的最大离子m/z191和m/z83得到两个化合物的分布图[/align]********************************************************************************[b]2. 利用二氢茉莉酮酸甲酯质谱单一离子面积计算估计化合物面积的方法来进行定量[/b]找到一个含二氢茉莉酮酸甲酯样品(可惜不是同一实验室的样品例子)来计算m/z83离子面积占化合物总面积的比例。[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/11/201911301122190332_1714_1615838_3.jpg!w690x387.jpg[/img][align=center]图6 二氢茉莉酮酸甲酯质谱图[/align]***********************************************************************************积分二氢茉莉酮酸甲酯总面积为45224507,如下。peak R.T. first max last PK peak corr. corr. % of # min scan scan scan TY height area % max. total26 32.534 6513 6658 6683 BV 385657 [b][u]45224507[/u][/b] 100.00% 13.064%积分m/z83离子的面积为10622419(仅计算反式异构体,顺式异构体在后面,保留时间不同)。Ion 83.00 (82.70 to 83.70): 20191123-1.D\data.msPeak # RetTimeType Width AreaStartTime End Time1 32.546 BB 0.136 [b][u]10622419[/u][/b] 32.144 32.6572 33.317 BB 0.067 1234446 33.146 33.464可以看出m/z 83离子积分面积占总面积的比例为:10622419/45224507*100=23.88%对未分离网友的溢隆香水样品的m/z83离子积分,结果如下:Ion 83.00 (82.70 to 83.70): 28.D\data.ms溢隆香水Peak # RetTimeType Width AreaStartTime End Time7 36.912 BV 0.144 [b][u]155890252[/u][/b] 35.860 37.024即二氢茉莉酮酸甲酯的大致面积为:155890252/23.88%=652806750积分未分离样品36.91分钟色谱峰,得到总面积为2953123137(所有离子积分面积)。peak R.T. first max last PK peak corr. corr. % of # min scan scan scan TY height area % max. total --- ----- ----- ---- ---- --- ------- ------- ------ -------88 36.911 5640 5746 5765 VV 511550053 [b][u]2953123137[/u][/b]100.00% 23.603%即ISO E Super的面积为:2953123137-652806750=2300316387即二氢茉莉酮酸甲酯:龙涎酮的大致面积比例为:652806750:2300316387 = 1:3.524[b]3. 利用龙涎酮质谱单一离子面积计算估计化合物面积的方法来进行定量[/b]同样可以利用iso E super龙涎酮的m/z191离子来估计估算龙涎酮的面积。找到另一个含龙涎酮的样品,此龙涎酮不是很大,这样可能会引起误差的,当然最好是同一实验室龙涎酮含量适中的例子,可惜我手头没有。[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/11/201911301124415112_3644_1615838_3.jpg!w690x387.jpg[/img][align=center]图7 含龙涎酮样品色谱图和质谱图[/align]**************************************************************************对m/z191离子进行积分,得到面积值为51231426,如下。Ion 191.00 (190.70 to 191.70):20191028.D\data.msSPPeak # RetTimeType Width AreaStartTime End Time3 62.225 PV 0.062 51231426 62.107 62.389对龙涎酮所有离子积分,即总面积为296362852,如下。peak R.T. first max last PK peak corr. corr. % of # min scan scan scan TY height area % max. total --- ----- ----- ---- ---- --- ------- ------- ------ -------76 62.225 14651 14671 14698 VV 6893554 296362852 21.20% 3.090%可以看出m/z 191离子积分面积占总面积的比例为:51231426/296362852*100=17.288%再看看未分离样品的情况:对m/z191进行积分。Ion 191.00 (190.70 to 191.70): 28.D\data.ms溢隆香水Peak # RetTimeType Width AreaStartTime End Time8 36.740 VV 0.305 [b][u]34031891[/u]0[/b] 36.271 36.9279 36.978 VV 0.071 [b][u]69389987[/u][/b] 36.927 37.024可以看到是两个面积,这是因为原来36.91分钟的m/z191离子分布如下(注意m/z83是单峰,请对照图5):[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/11/201911301125343462_3626_1615838_3.jpg!w690x387.jpg[/img][align=center]图8 样品36.91分钟的m/z191离子分布[/align]**********************************************************************************即m/z191离子积分面积为340318910+69389987=409708897409708897/17.288%=2369903384和2300316387相差不大,可以接受。由于这两个二氢茉莉酮酸甲酯和龙涎酮的纯净质谱图均来自别的网友的数据,不是原来样品网友的数据,因为不同调谐参数和分析条件会引起误差的。如果使用自己在相同条件的香料样品测定得到的质谱图,估算会更准确。
求:软脂酸的核磁共振氢谱图
用盐酸或盐酸加硝酸溶解稀土硅镁合金最后溶解都不太完全,由于要用ICP测硅,不能加氢氟酸,请问用何种方法能完全溶解稀土硅镁合金。
各位大哥,在沒有塑料燒杯的情況下,怎麽趕走氫氟酸阿?我們公司的ICP霧化室耐不了氫氟酸的阿! 救救!
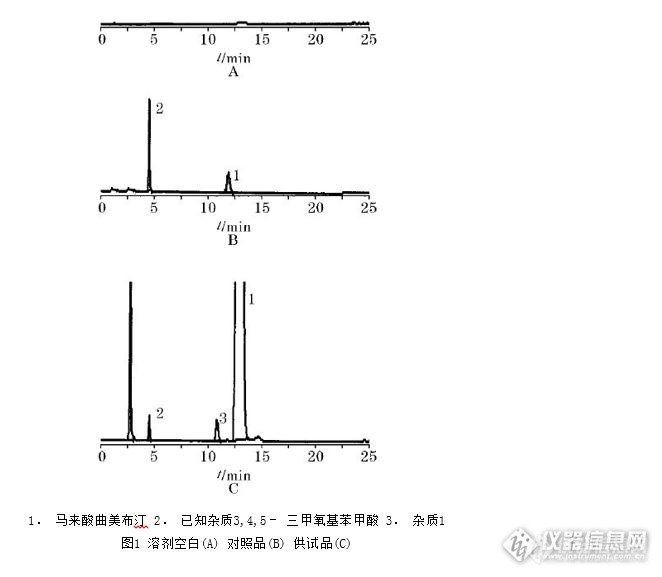
马来酸曲美布汀为白色结晶或结晶性粉末,无臭,味苦。在冰酯酸和氯仿中易溶,在乙腈和甲醇中溶解,在水和无水乙醇中微溶;在乙醚中几乎不溶。其作用与用途是胃肠道运动功能紊乱引起的食欲不振、恶心、呕吐、嗳气、腹胀、腹鸣、腹痛、腹泻便秘等症状的改善以及肠道易激惹综合征。以下为使用资生堂C18色谱柱对马来酸曲美布汀检测得到的谱图,请参考。http://ng1.17img.cn/bbsfiles/images/2016/11/201611030956_615667_0_3.jpg色谱条件色谱柱:CAPCELL PAK C18 S5; 4.6 mm i.d.×250 mm流动相:缓冲液(取高氯酸0.43mL,加水950mL,混匀后用醋酸铵溶液调节pH值至(3.75±0.05),用水稀释至1000mL,加戊烷磺酸钠1.54g振摇使溶解)/乙腈=65/35流 速 : 1.0mL/min温 度 :40℃检 测 : UV 268nm进样量:20μL
如题:寻找硫酸双肼屈嗪的紫外谱图。谢谢了。希望有高人帮助。
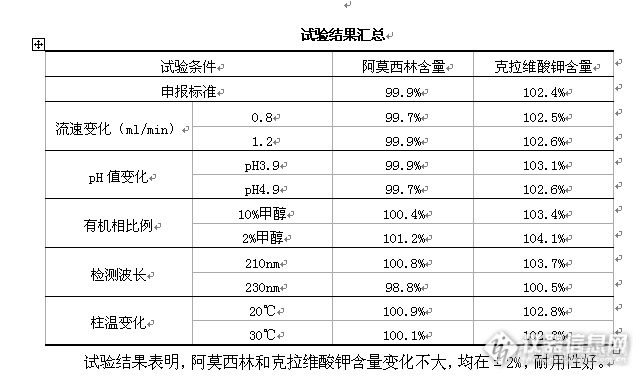
继”阿莫西林克拉维酸钾胶囊有关物质方法学”项目结束,整理的含量测定方法学。项目:含量测定(3.2.P.5.2.9)检查方法:照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定试验条件:仪器:LC-2010CHT (SHIMADZU)万分之一电子天平(Sartorius ABS-124S型)工作站(LCsolution色谱工作站)色谱柱(填料:C18,规格:250mm×4.6mm,填料粒径:5μm)Xtimate C18 4.6*250 ,PN:Xt5B18425 ,SN:411101950UV检测器(检测波长:220nm)柱温:室温流动相:0.05mol/L磷酸二氢钠溶液(取磷酸二氢钠7.8g,加水900ml使溶解,用10%磷酸溶液或氢氧化钠试液调节pH值至4.4±0.1,加水稀释至1000ml)-甲醇(95:5)。流速:1.0ml/min。运行时间:约20分钟。系统适用性:取阿莫西林克拉维酸系统适用性试验对照品,加流动相溶解并稀释制成每1ml中含0.8mg的溶液,取20μl注入液相色谱仪,记录的色谱图应与标准图谱一致。具体试验操作:取装量差异项下的内容物适量,精密称取适量,加水适量,超声使溶解并定量稀释制成每1ml中含阿莫西林0.5mg的溶液,滤过,立即精密量取续滤液20μl注入液相色谱仪,记录色谱图;另分别精密称取阿莫西林对照品与克拉维酸对照品各适量,加水溶解并定量稀释制成每1ml中约含阿莫西林0.5mg和每1ml中含克拉维酸0.125mg的混合溶液,同法测定。按外标法以峰面积分别计算供试品中C16H19N3O5S和C8H9NO5的含量。计算公式:标示量百分含量(%)=××100%式中:Cs为对照品的浓度(mg/ml);At为供试液的主峰面积;Nt为供试液的稀释倍数;AS为对照品溶液的主峰面积;W为供试品取样量(mg)。“色”路蹒跚,藤下葡萄,某品种含量测定方法学之耐用性试验部分路蹒跚,藤下葡萄,某品种含量测定方法学之耐用性试验部分http://ng1.17img.cn/bbsfiles/images/2013/06/201306292159_448382_1621890_3.gif3.2.P.5.3.6.1波长选择本品含量测定检测波长参照中国药典2010年版二部收载的阿莫西林克拉维酸钾相关制剂质量标准含量测定项,即220nm。3.2.P.5.3.6.2流动相选择(色谱图见附件1122~1124)参照中国药典2010年版二部收载的阿莫西林克拉维酸钾相关制剂质量标准含量测定项,以0.05mol/L磷酸二氢钠溶液(取磷酸二氢钠7.8g,加水900ml使溶解,用10%磷酸溶液或氢氧化钠试液调节pH值至4.4±0.1,加水稀释至1000ml)-甲醇(95:5)为流动相。试验过程:系统适用性试验供试液:精密称取阿莫西林克拉维酸钾系统适用性对照品4.2mg至5ml量瓶中,加流动相适量超声使溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试液;对照品溶液:精密称取阿莫西林对照品29.1mg和克拉维酸钾对照品7.3mg至50ml量瓶中,加水适量超声使溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试液;精密量取上述供试液各20μl注入高效液相色谱仪,记录色谱图,典型色谱图见下图:http://ng1.17img.cn/bbsfiles/images/2013/06/201306292200_448383_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292200_448384_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292201_448385_1621890_3.gif3.2.P.5.3.6.3进样精密度试验(色谱图见附件1125~1130)
用石墨消解仪消解标准土壤,消解过程如下:取0.5g标土于聚四氟乙烯消解管中,加少量水润湿,加10毫升浓盐酸摇匀,120度消解一个半小时,液体差不多剩余1毫升;取下消解管,稍冷,加5毫升硝酸,5毫升氢氟酸,3毫升高氯酸,摇匀,加盖,150度消解两个小时,取下盖子,150度消解一个小时;升温至180度赶酸一个小时,状态为粘稠不易流动;取下稍冷,加水,加1毫升浓硝酸,定容至50毫升容量瓶中,静置24小时,取上层清液上机测试。经过上述过程消解后的标土上机测试,火焰法测Cu,Ni,Zn,总Cr,等测量值都在标准范围内,石墨炉测Pb,测定值也在标准范围内,只有石墨炉测Cd,测量值只有标准值的70%,偏小很多。请各位专家帮我分析分析是哪里出了问题,谢谢。
各位大侠:请帮我在你的谱库中搜索一下去氢木香内酯的质谱图,帮忙查一下它的定性离子!万分感谢!

10,抽取5个版友);中奖名单:WUYUWUQIU(注册ID:wulin321)莫名其妙(注册ID:moyueqiu)玲儿响叮当(注册ID:jshbhh)千层峰(注册ID:jxyan)吕梁山(注册ID:shih20j07)http://ng1.17img.cn/bbsfiles/images/2016/12/201612141537_01_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612141537_02_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================苯甲酸、山梨酸、糖精钠、安赛蜜、脱氢乙酸的测定方法:HPLC基质:标准溶液应用编号:101812化合物:苯甲酸、山梨酸、糖精钠、安赛蜜、脱氢乙酸固定相:Diamonsil C18(2)色谱柱/前处理小柱:Diamonsil 5μm C18(2), 250 x 4.6mm样品前处理:样品制备 制备方法: 分别取苯甲酸、山梨酸、糖精钠、安赛蜜、脱氢乙酸适量,用水溶解,配成浓度为0.02 mg/mL的混合标准溶液。色谱条件:分析条件 色谱柱: Diamonsil C18(2),250×4.6 mm,5 μm (Cat#:99603) 流动相: 甲醇:0.02 mol/L乙酸铵=6:94 流速: 1.0 mL/min 柱温: 30 ℃ 检测器: UV 230 nm 进样量: 10 μL文章出处:天津迪马实验室关键字:苯甲酸,山梨酸,糖精钠,安赛蜜,脱氢乙酸,Diamonsil C18(2),99603摘要:苯甲酸、山梨酸、糖精钠、安赛蜜、脱氢乙酸的测定谱图:http://www.dikma.com.cn/Public/Uploads/images/ansaimi3(1).PNG备注:1-安赛蜜 2-苯甲酸 3-山梨酸 4-糖精钠 5-脱氢乙酸

这个是二氢茉莉酮酸甲酯吗http://ng1.17img.cn/bbsfiles/images/2015/06/201506111120_549832_2015686_3.jpg
[align=center][size=24px]一次饮料中脱氢乙酸的测定思考[/size][/align] [size=20px]饮料中脱氢乙酸,按照GB5009.121—2016,流动相是甲醇/0.02mol/L乙酸铵(10/90)体系,在此体系下,如果不是新色谱柱,脱氢乙酸峰型不好,容易拖尾,如图:1,[/size] 图1: [img]https://ng1.17img.cn/bbsfiles/images/2024/08/202408161621223084_8858_5326750_3.png[/img] [size=20px]为迎接质控考核,想解决脱氢乙酸峰型不好的问题。在通过咨询老师同事,考虑使用甲醇/0.1%磷酸水体系,在50/50等度洗脱下,脱氢乙酸的出峰时间提前,峰型得到较大改善,响应值也得到明显提升,如图:2[/size] [size=20px]图2:[/size] [img]https://ng1.17img.cn/bbsfiles/images/2024/08/202408161621224268_4374_5326750_3.png[/img] [size=20px]在以为万事大吉,可以放心做质控了,随采用甲醇/0.1%磷酸水体系,在50/50等度洗脱下,对考核样品进行测定:标准溶液为脱氢乙酸单标,盲样为橙汁中的脱氢乙酸,购买质控样品为:苹果汁、桑葚汁进行测定,结果质控品测定值均不太理想,尤其是桑椹汁,结果高出5倍。尝试优化流动相比例,改为65/35(甲醇/0.1%磷酸水)洗脱,发现桑椹汁出现两个峰,判断桑椹汁质控品,应该还有山梨酸或苯甲酸等物质,如图3:[/size] 图3: [img]https://ng1.17img.cn/bbsfiles/images/2024/08/202408161621229981_532_5326750_3.png[/img] [size=20px]确定购买的质控品可能含有其他防腐剂,故仍改用甲醇/0.02mol/L乙酸铵(10/90)体系,并用山梨酸、苯甲酸、脱氢乙酸混合标准测试,并调整柱温后,拖尾有稍许改善,也可能是使用甲醇/0.1%磷酸水过程中,对柱子有修复作用。混合标准图谱见图4:桑椹汁图谱见图5,甲醇/0.02mol/L乙酸铵(10/90)体系,对三种防腐剂有较好的分离,质控样中杂质已很好的分开,脱氢乙酸拖尾不严重。[/size] [size=20px]图4:[/size] [img]https://ng1.17img.cn/bbsfiles/images/2024/08/202408161621227271_2806_5326750_3.png[/img] [size=20px]图5:[/size] [img]https://ng1.17img.cn/bbsfiles/images/2024/08/202408161621228456_3546_5326750_3.png[/img][size=20px]重新配制标准曲线、处理盲样、质控品,进行测定,得到满意结果。[/size] [size=20px]回顾总结:1、脱氢乙酸在甲醇/乙酸铵体系下拖尾较易发生,尤其测定大批量样品后,脱氢乙酸峰拖尾会非常严重,2、升高柱温会对拖尾峰型有所改善、使用酸体系流动相后色谱柱有明显的修复改善,3、甲醇/0.1%磷酸水体系下,脱氢乙酸峰型、响应非常好,但该体系下可能对饮料中常见防腐剂等物质分离不够理想,如本案例中山梨酸和苯甲酸,应该设置梯度洗脱实现分离,如果能[/size][size=20px]确认盲样和质控品只含脱氢乙酸的情况下,甲醇/0.1%磷酸水体系是一个很好的替代方案。[/size]
苦味酸溶液测煤气中萘含量的,国标指标名用一瓶25克的苦味酸溶于2000ml水中。煮沸,冷却后过滤,取上清液用0.1mol/L氢氧化钠滴定配成0.042mol/L的吸收溶液。不知道用多少毫升滴定到何种状态,请求老师们告知。
各位,请问用0.2mol/L的三氟乙酸去测定硫酸庆大霉素的时候,0.2mol/L的三氟乙酸pH0.58左右,而我用的C18柱子最低能耐受pH为1,请问各位0.2mol/L的三氟乙酸需要调节pH吗,如果需要用什么调节
有人测过碳酸氢钠粉末的激光粒度吗?用什么方法测试的?
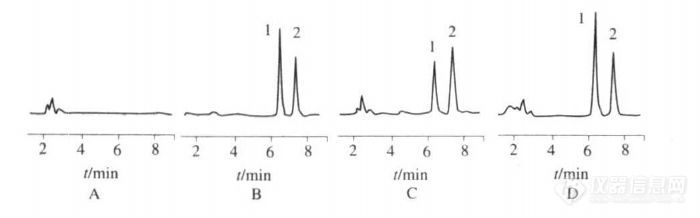
【作者】 王彦慧; 夏东亚; 郭涛;【机构】 沈阳军区总医院药剂科; 沈阳军区总医院药剂科 辽宁沈阳110016; 沈阳药科大学; 辽宁沈阳110016;【摘要】 目的:建立测定人血浆中盐酸曲马多的高效液相色谱-荧光检测法。方法:血浆样品经液-液萃取,以氟康唑为内标,在Diamonsil C18柱(迪马公司,200mm×4.6mm,5μm),以磷酸二氢纳缓冲液(0.04mol.L-1,pH3.5)-乙腈(77∶23)为流动相,荧光检测,激发波长为275nm,发射波长为302nm。结果:曲马多的线性范围为5~1 000μg.L-1,最低检测限为1μg.L-1。低、中、高3种浓度的日内日间RSD均小于7.7%,提取回收率分别为(85.2±4.3)%,(80.6±3.5)%和(83.6±3.6)%。结论:该法重复性好、准确、简便、快速,适用于人血浆中盐酸曲马多浓度的测定及临床药动学研究。 【谱图】 http://ng1.17img.cn/bbsfiles/images/2012/08/201208142231_383915_1609970_3.jpg
氢氟酸要用哪些器具取?必须用聚四乙烯容器来消解吗?
在办公室里做事,突然听到分析室里有人在大叫了一下,我飞驰过去,我们一位分析人员手捧着手不知所措,痛苦表情让我知道手上一定沾上化学试剂了,顺手拿了一个干毛巾,在他手上沾擦后,马上拉他的手在水龙头下冲洗。一分钟后他的表情稍稍轻松下来,问他触到什么了?他指了指桌台上的抹布说,我准备用它来搽烧杯,刚用手拿起来准备用,就感到手上发热,刺痛.....我让他自己手放在水龙头下继续冲,然后小心检查抹布:那是一块湿抹布,里面有好几块洞,同时还可以看到烟雾在冒。这时另一位同事从另外一个分析室里出来说:上面是硫酸,刚才我在溶样,不小心把硫酸撒出来一些,马上戴上防酸碱手套,用湿抹布对工作台进行了擦拭,另外一间分析室里有事就把抹布放在水池里,想等回来就冲洗... 我晕,把他训了顿,然后把被烧伤的同事手拿起来看看,好几块红点,不知会不会起泡。让他再冲一会,再给他上点烧伤膏,准备让他先回去休息一下啦。请大家引事发表看法,如何让我们身边减少类似危险!
在采用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定脱氢乙酸时,第一组的标曲中,没有杂峰但是低浓度目标峰的峰面积普遍偏低,再测第二组时,标曲普遍含有很多杂峰,目前已经采取过烘柱子来去除柱子中的杂质,也更换过衬管,上述情况依旧存在,请教各位前辈老师,是否遇到过类似情况,帮忙解答,谢谢![url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]型号:安捷伦7890B[img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307050929527429_8238_5640885_3.png[/img]
各位大神,求指点!做脱氢乙酸有情况。按GB/T23377做的,标样很好,线性6个9,但是样品加标回收低,只有50-60%。流动相PH大概是6-7,0.02mol乙酸铵:甲醇=90:10。做了两组样品,一组是饮料加标,样品离心后没有过萃取小柱,样品PH调成4-5一组是面包加标,样品用亚铁氰化钾和乙酸锌除蛋白,样品离心后用正己烷萃取除脂肪,样品PH调成4-5加标的结果不对啊,饮料加标25ug定容到25ml,看不出什么峰形,几乎检不出来,加标600ug定容到25ml,回收率只有63.75%面包加标25ug定容到25ml,几乎检不出来,加标600ug定容到25ml,回收率只有55%目标峰有拖尾,但不至于这么影响回收率吧?!
误食食品级盐酸或者过氧化氢等不能催吐吗?误食氢氧化钠可以催吐?禁止催吐的原因是什么?
在很长的一段时间内,人们认为王水就是酸中之王,是最强的酸了,因为即使是黄金,遇到王水也会像“泥牛入海”一样很快变的无影无踪。 直到有一天奥莱教授和他的学生偶然发现了一种奇特的溶液,它能溶解不溶于王水的高级烷烃蜡烛,人们才知道其实王水并不是最强的酸,还有比它强的酸,这就是魔酸,又叫超强酸。 从成分上看,超强酸是由两种或两种以上的含氟化合物组成的溶液。它们的酸性强的令人难以置信,比如氢氟酸和五氟化铅按1 :0.3(摩尔比)混合时,它的酸性是浓硫酸的1亿倍;按1 :1混合时,它的酸性是浓硫酸的10亿倍。所以王水在它们面前只能是“小巫见大巫”。 由于超强酸的酸性和腐蚀性强的出奇,所以过去一些极难或根本无法实现的化学反应,在超强酸的条件下便能顺利进行。比如正丁烷,在超强酸的作用下,可以发生碳氢键的断裂,生成氢气,也可以发生碳键的断裂,生成甲烷,还可以发生异构化生成异丁烷,这些都是普通酸做不到的。 自从奥莱教授和他的学生发现超强酸以后,人们又开始研究起强酸,可以说是他们重新点燃了人们研究强酸的兴趣。迄今为止,化学家已找到了多种新的超强酸。也许在不久的将来还会发现更多的超强酸。 现在已知的几种超强酸,除了可以做酸性催化剂外,还可以做有机化合物或无机化合物的质子化剂,至于在其他领域有没有应用还等待人们去发现。
本人遇到铝合金中含锆、稀土(铈和镧)、铁、硅等元素,用分光光度法或EDTA滴定法测试锆无法测试准确,因稀土严重干扰锆的测定。据资料介绍1)分离钍、稀土:将含锆的稀土沉淀于热浓硫酸(或高氯氯酸和硝酸)中,然后调节酸度进行草酸盐沉淀,全部锆均可留在溶液中。问题: 1、称取试样(铝基材)量为多少。 2、取多少ml酸酸,温度是多少适宜。 3、调节酸度为多少mol/L为宜。 4、取用哪种草酸盐,用量多少可将锆与稀土分离。2)在10mol/L盐酸介质中,用苯甲酰苯胲萃取锆,可与铀、钍、稀土、铌、钽、钛等分离。有机相中的锆以氢氟酸——盐酸反萃取至水相中,可用光度法测定。问题: 1、称取试样(铝基材)量为多少。 2、苯甲酰苯胲萃浓度是多少为宜 3、氢氟酸——盐酸各自浓度为多少以多少比例混合。请高手指点,还是有更合好的分离方法或具体的测试同时含有稀土和锆的方法(由于实验条件,只能局限于分光光度分和滴定法),谢谢!
各位,请问用0.2mol/L的三氟乙酸去测定硫酸庆大霉素的时候,0.2mol/L的三氟乙酸pH0.58左右,而我用的C18柱子最低能耐受pH为1,请问各位0.2mol/L的三氟乙酸需要调节pH吗,如果需要用什么调节







 我要推广仪器
我要推广仪器
 下载APP
下载APP