我用LC-MS 测定雌二醇、雌三醇、雌酮 ,在摸索质谱条件时 ,总找不到碎片离子 ,ESI+和ESI-都试过 参考文献用别人的质谱条件 ,做液质联用时不出峰 ,别人用的是C18的柱子 ,我们用的是C8 是柱子的问题吗 ?还是质谱的问题 ?我应该怎么解决呢
液相测雌三醇等7种性激素,一套有证标准物质下来多少钱?都有用哪些品牌?质控CRM怎么选择?
GB 29698-2013 食品安全国家标准 奶及奶制品中17β-雌二醇、雌三醇、炔雌醇多残留的测定 气相色谱-质谱法
请教大家,我的反应物进样GC-MS的时候显示有三个杂质,我的反应物是 4-chlorobiphenyl这三个杂质分别是 biphenyl ; 3-ethyl biphenyl; 4,4'-dichlorobiphenyl。为了不影响实验结果,我想想办法把这三个杂质去掉,尽量再提纯,虽然反应物买来的时候供应商说他们的纯度98.8%,但随着储存的时间长,貌似纯度越来越低了,以至于实验完了我没法判断我的产物是来自我的反应物还是这些杂质。请问大家有什么好方法来提纯我的反应物吗? 这几个物质结构相近,沸点也相差没有那么多。 虽然这个提纯的知识点高中就学了,但看了一遍竟然觉得没有一个可操作性强的……先行谢过各位大侠!!
紧急求助各位论坛前辈: 我们公司在用甲醇溶解水飞蓟素进行紫外法含量的测定时,出现了OOS结果,在调查过程中发现使用分析纯甲醇溶解水飞蓟素时颜色比使用色谱纯溶解时颜色深,因此怀疑是分析纯甲醇中含有某种杂质与水飞蓟素反应引入显色基团而导致水飞蓟素颜色加深导致测定结果偏高。 我想请教那位仁兄知道甲醇的杂质检查方法,希望能够发来交流一下。 我已经初步做了一点工作,是按照中国药典2005版的溶剂残留法按面积归一化法检查的,但由于在这里甲醇是主要物质,使用此方法不太合适,因此没有检验出来分析纯含有何种杂质比色谱纯中的高。 另外有那位仁兄知道天津永大化学试剂厂质控部的电话,希望可以告诉一下,有在那里工作的大家一起交流一下更好。我的QQ是13107914。谢谢
药物杂质中有三甲胺、溴代十六烷和溴代十八烷(沸点超过300度)。其中三甲胺标准品的溶剂为甲醇,浓度为100mg/L。做药物杂质的含量分析时,如用DB-5ms柱,进样口温度为310度,则柱温25度甲醇峰掩盖三甲胺的峰;溴代十六烷和溴代十八烷在220度保持20分钟会出峰。如用DB-wax柱,进样口温度为240度,溶剂为乙醚。如何证明溴代十六烷和溴代十八烷在进样口中完全气化。

在甲基培尼皮质醇的制造过程中,可能会生成一些杂质。这些杂质可能会出现在原料中,也可能在制药过程中的化学反应中产生。不论其来源,杂质的存在都可能影响到药物的质量、安全性和疗效。例如,甲基培尼皮质杂质可能增加药物的毒性,或导致不良反应。同样,杂质也可能对甲基培尼皮质醇的药效产生影响。因此,制药公司必须在生产过程中严格检测和控制这些杂质。检测和控制药品中的杂质是药品质量控制的重要组成部分。CATO标准品对杂质的研究不仅有助于保证药品的质量和安全性,也可以为优化制药过程提供参考。比如,通过对杂质的研究,可以找到产生这些杂质的原因,从而改进制药过程,减少杂质的生成。[img=,600,588]https://ng1.17img.cn/bbsfiles/images/2024/02/202402052058222428_1748_6381668_3.png!w600x588.jpg[/img]
丁醇产品中除了有异丁醇、辛醇异戊醇等杂质外,还可能有哪些杂质?
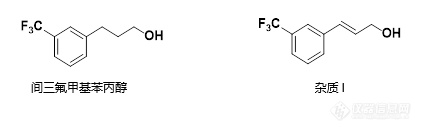
[align=center][b]间三氟甲基苯丙醇和杂质I的分离[/b][/align]客户提供了间三氟甲基苯丙醇和相关杂质I,并反馈曾尝试使用反相C[sub]18[/sub]柱对两化合物进行分离,但未能得到基线分离结果。现客户希望本实验室选择合适色谱柱并对色谱条件进行优化,来实现间氟甲基苯丙醇和其相关杂质I的基线分离。首先,我们尝试使用中等极性的CAPCELLPAK C[sub]18[/sub] MGII色谱柱,在磷酸盐-乙腈体系中分析50 μg/mL的混标溶液及各单标溶液,通过调整流动相中水相和有机相比例为60:40时,50 μg/mL的混标溶液中,间三氟甲基苯丙醇和杂质I能实现基线分离,分离度为1.52(见图1)。同客户沟通,客户希望供试品溶液(当间三氟甲基苯丙醇浓度为1mg/mL,杂质I为1 μg/mL)中两化合物分离度大于1.50。[align=center][img=,422,132]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009027392_4941_2222981_3.png!w422x132.jpg[/img][/align][align=center][img=,656,427]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009243004_918_2222981_3.png!w656x427.jpg[/img][/align][align=center]图1 MGII分析混标及单标溶液结果[/align][align=left][img=,575,197]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009245664_7431_2222981_3.png!w575x197.jpg[/img][/align][align=left]在此实验基础上,进一步分析供试品溶液,结果发现由于间三氟甲基苯丙醇浓度过高,致使色谱峰展宽,杂质I与间三氟甲基苯丙醇的分离度下降,未能达到1.50的基线分离要求;进一步尝试通过升高柱温来改善分离度,结果如图2,在50°C时能够得到良好分离结果,分离度为1.59。[/align][align=left][/align][align=center][img=,650,418]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031030364182_5088_2222981_3.png!w650x418.jpg[/img][/align][align=center]图2 MGII分析混标及单标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,575,195]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031031319132_5141_2222981_3.png!w575x195.jpg[/img][/align][align=left][/align][align=left]为有更多的选择,我们也尝试了两款非C[sub]18[/sub]色谱柱,包括键合特殊官能团——金刚烷基的高极性色谱柱ADME和键合五氟苯基的PFP色谱柱。在使用PFP色谱柱分析50 μg/mL混标溶液时,发现两化合物峰重合,未能实现分离。但使用ADME分析混标溶液时,能够得到1.36的分离度(见图3)。[/align][align=left][/align][align=center][img=,620,423]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034384978_3594_2222981_3.png!w620x423.jpg[/img][/align][align=center]图3 PFP、ADME分析50 μg/mL混标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,552,214]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034366042_2199_2222981_3.png!w552x214.jpg[/img][/align][align=left][/align][align=left]尝试改善分离度,继续使用ADME色谱柱进行分析,通过降低有机相比例来延长保留,最终得到了1.50的分离度(见图4),与此同时对供试品溶液进行分析,发现由于主成分峰展宽未能得到基线分离结果(见图5)。[/align][align=left][/align][align=center][img=,658,430]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035399180_5905_2222981_3.png!w658x430.jpg[/img][/align][align=center]图4 ADME分析混标溶液结果[/align][align=center][/align][align=center][img=,657,435]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035148034_8911_2222981_3.png!w657x435.jpg[/img][/align][align=center]图5 ADME分析供试品溶液结果[/align]注: 峰上标数字为分离度。[align=left][img=,586,223]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035150115_8050_2222981_3.png!w586x223.jpg[/img][/align]
老板要我检测原料的纯度,因为如果杂质含量过高会影响产品质量,所以还需要检测杂质的含量,而杂质没有标样,我可怎么定量呀. 杂质为 2,4-双酚A,色满和三酚,哪位专家帮帮忙吧
做乙醇的挥发性杂质的时候 乙醛和甲醇的分离度 很难调 求助,然后我用相同的色谱条件 相同的柱子 第一次进能分开,第二次进就分不开 这是为何?
本人在工作中遇到一个难题,就是有一批回收甲醇要处理,要检测甲醇的含量和个杂质的含量,但是目前只有原料样品和最终产品样品,中间体什么的,或者副产物什么的标准品没有,我走[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]只有甲醇的峰,但是走液相大概有十几个峰,请问各位大侠,我该如何检测呢?是不是只有过柱子提纯然后做质谱,核磁什么的才能确定呢?其中杂质不是产品,原料还没有确定,但是原料只有4种。杂质有十几种,请高人给指条路[em0808]
请问各位大峡 甲醇中初水之外只要含有什么杂质,是什么东西能使高锰酸钾褪色?
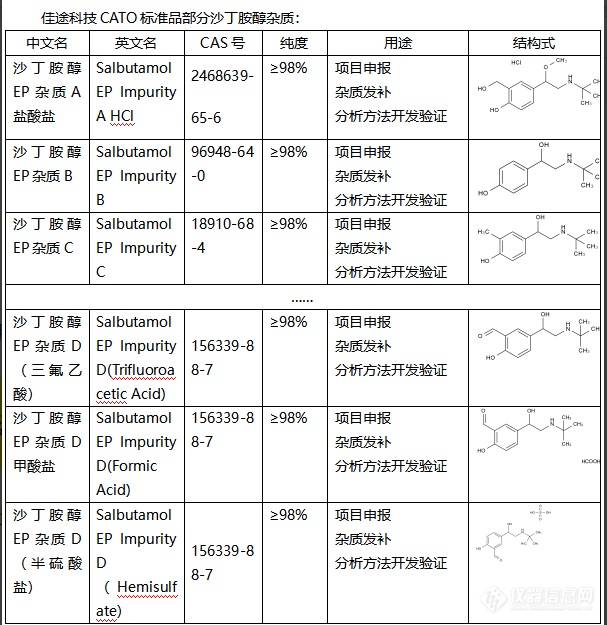
沙丁胺醇是一种药物,主要用于治疗哮喘和慢性阻塞性肺病等呼吸系统疾病。在药品制备过程中,可能会产生一些不同结构的化合物,这些化合物就被称为杂质。杂质可能会对药品的品质、安全性、效力和稳定性等产生影响。根据产生的差异,杂质的作用可以具体表现如下:1. 影响药品的稳定性:杂质可能导致药品在贮存过程中发生化学变化,从而影响药品的稳定性。2. 影响药品的安全性:如果杂质对人体有毒性,那么杂质的存在可能降低药品的安全性。3. 影响药品的效力:杂质可能与药品的有效成分发生竞争,导致药品的效力降低。4. 影响药品的品质:杂质可能改变药品的外观、颜色、溶解性等物理性质,影响药品的品质。CATO标准品制药行业对药品的杂质进行严格控制,保障药品的质量和患者的用药安全。[img=,607,625]https://ng1.17img.cn/bbsfiles/images/2024/02/202402021631332927_8650_6381668_3.png!w607x625.jpg[/img]
反应物为烷基咪唑和卤代酯反应生成离子液体,在氢谱中,都有3.3或3.9的杂质峰,0.8,0.9,1.2的杂质峰,量都很少。我想1.2可能是硅胶的杂质,1.2和3.3可能是乙醚的杂质,但是柱层析下来的蒸后应该没有乙醚了吧。3.3也可能是甲醇,因为酯化反应可能会有一部分醇生成,醇在离子液体中溶解性又很好,很难除去。可是烷基咪唑的核磁谱中也有3.3的蜂,这我就不明白了,会是什么杂质呢,还有资3.8、3.9的小杂质峰会是什么物质呢?请教各位大侠帮帮忙解释一下吧,先谢谢了。
第一次做乙醇挥发性杂质,仪器是岛津2014C,柱子安捷伦DB-624,30*0.32*1.8um,甲醇,乙醛,乙缩醛,苯都是用色谱纯级别的,按照药典,进对照a、b、c,d,都没有峰,只有一个大的乙醇峰,我进的是1ul,采用分流进样,流速是2ml/mian,做不出来,在这里请问各位有经验的老师,怎么设置参数?-----------做的我快崩溃了,在这里先谢谢各位老师!
我是一个新手,我做乙醇挥发性杂质时,我们的样品里面均未检出各杂质,是否合理?怎么感觉多少都应该有点呢?还是因为我是放置了几天以后才做样的缘故?请教各位!谢谢谢谢!
供试品溶液(b)中其他各杂质峰面积的总和不得大于4-甲基-2-戊醇的峰面积(0.03%,以4-甲基-2-戊醇计)。这个其他各杂质峰包不包括甲醇、乙醛、乙缩醛和苯?求指点!
各位谁有关于甲醇中杂质含量测定,是关于工业生产甲醇的,什么方法都可以,我查到一篇文献,但是要收费,我看不了,是《[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定甲醇中杂质含量》,谢谢各位了
我是第一次做乙醇中挥发性杂质的检测,按照药典条件,进了1ul的对照a b c d, 出来的就只有一个大的乙醇峰,甲醇,乙醛,乙缩醛,苯,这四个成分都没有峰出来,这是什么原因,请各位老师,帮帮忙,谢谢
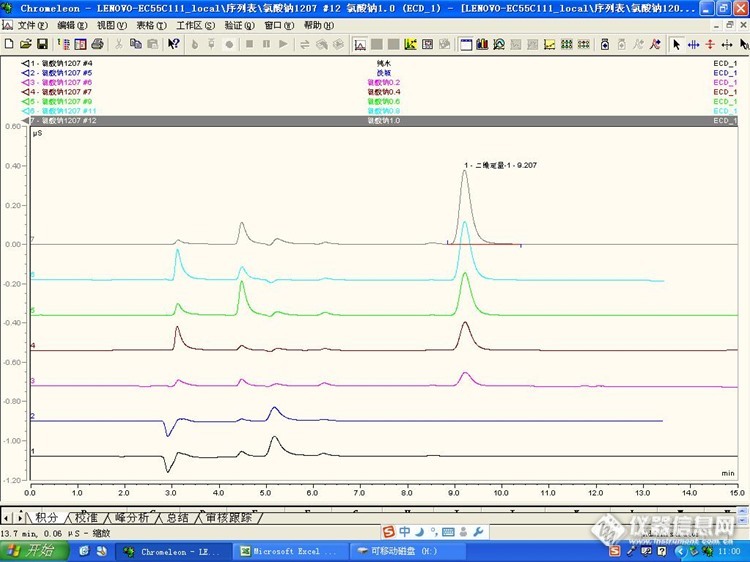
1、开始进三次超纯水有重现性并且没有杂质峰2、进样品,样品峰重现性很好,但是杂质峰没有规律忽大忽小3、想问一下水负峰后紧跟的峰大概是什么?图片是扣除空白之后的http://ng1.17img.cn/bbsfiles/images/2016/12/201612120909_01_3088211_3.jpg
“杂质度过滤板”是在乳品检测过程中用来测定乳品杂质含量的一种专用产品,其质量的好坏直接关系到检测结果的准确性与权威性。近几年来,在乳品检测仪器市场上出现了许多仿冒、贴牌的伪劣“杂质度过滤板”产品,对乳品检测结果的准确性与公正性造成了危害,由此,记者对“杂质度过滤板”专利的发明人李世春,就该产品的研究发明过程、国家标准的修改制定、专利权保护以及市场环节等相关问题进行了采访。 记者:杂质度过滤板在乳品质量检测过程中的作用是怎样的? 李世春:所谓杂质度过滤板,其实是一个很小的产品,主要是用来测定乳、乳粉类产品杂质度项目中不可缺少的一种专用过滤产品,它本身是否合格、标准,直接影响到被检测乳品杂质度结果的准确性。 记者:为什么说 “杂质度过滤板”是填补了我国同类产品空白的一项发明? 李世春:因为早在1990年以前我国乳品检验中没有杂质度过滤板这个专有名词和这个专用产品。国家标准(GB5413-85.2.7)中对杂质度的测定也只有“棉质过滤板:直径32mm,过滤时牛乳通过直径为28.6mm圆”这么一句话,而这只是简单说了棉质过滤板的规格,却没有棉质过滤板本身的任何技术指标与技术要求。这使得我国乳品行业各单位在测定乳、乳粉类产品中使用的棉质过滤板五花八门、各不相同,其测定结果可想而知。 记者:当时国外同类产品是什么样的水平? 李世春:当时在国外,每个国家使用的过滤板也是各不相同。当时我还在国家乳制品质量监督检验中心理化检验室工作,决心改变当时我国测定乳、乳粉类产品杂质度项目无统一的专用过滤板现状,自1987年开始就研究试制测定乳、乳粉类产品杂质度项目的专用过滤板。经过5年多的尝试,最后在1992年试制成功,第二年获得了国家发明专利。随之国内各乳品企事业单位陆续使用,并受到好评。 记者:是不是由于“杂质度过滤板”这项发明在业内产生了一定的影响,因此而获得起草修订相关国家标准的任务? 李世春:不是,是在“杂质度过滤板”发明之前,黑龙江省乳品工业技术研究所根据国家轻工业部食品局的要求,责成我先后修订起草了国家标准GB5413-85.2.7杂质度的测定,同时,起草制作了国家标准GB5413-85彩色《杂质度标准板》(1984年),并发表 了《杂质度标准板》的使用说明论文。在此基础之上,又研制成功了测定乳、乳粉类产品杂质度过滤机,为此,在1993年申报该过滤板国家发明专利时,将其命名为“杂质度过滤板”,以此区别于国家标准中的“棉质过滤板”。从那以后“杂质度过滤板”就逐渐成为了我国在测定乳、乳粉类产品杂质度项目里的专有名词,也被业内人士广泛称呼使用。“杂质度过滤板”这一产品也成为了我国在测定乳、乳粉类产品杂质度项目的专用产品广为使用。 记者:“杂质度过滤板”这项发明最大的意义在于何处呢? 李世春:“杂质度过滤板”这项发明专利中明确了该产品的特征技术标准,如材质、制造工艺、密度、厚度、孔径、白度、尘埃度、透气度等。而这些特征技术标准是实现“杂质度过滤板”全国统一和保证测定杂质度项目结果准确性的前提条件。由于“杂质度过滤板”产品的市场化前景很好,国家乳制品质量监督检验中心还和我在1999年签定了“杂质度过滤板专利技术使用和该专利产品销售协议”。自1992年后的10余年中,应该说我国的杂质度过滤板基本上实现了专用和统一。 记者:据说现在市场上出现了很多仿冒和伪劣的“杂质度过滤板”产品? 李世春:是的,目前的国内化玻仪器市场上,尤其是哈尔滨市化玻仪器市场上出现了多家大量假冒伪劣的所谓“杂质度过滤板”,有的甚至直接声称是在我这里进的货。我对部分假冒伪劣的所谓“杂质度过滤板”分析检验后发现这些假冒伪劣的所谓“杂质度过滤板”除了材质和加工工艺外,其他绝大多数特征技术标准不符合我“杂质度过滤板” 国家发明专利的技术要求。这些假冒伪劣的所谓“杂质度过滤板”产品多数来自那些前些年曾在国家乳制品质量监督检验中心或其下属公司从事销售过该产品,但因销售部门解体而被分流或辞退的某些人员所办的公司;少数来自某些曾经经销过该产品的化玻仪器商店。如果这种情况继续下去,我国测定乳、乳粉类杂质度项目将倒退,对测定乳、 乳粉类产品杂质度项目的单位来说,选择了那些假冒伪劣的所谓“杂质度过滤板”,将无法保证测定杂质度项目的准确性。“杂质度过滤板”特征技术标准的统一也就无从谈起。 记者:“杂质度过滤板”特征技术标准是怎样的? 李世春:提起标准更令人遗憾。从上次修订后的国家标准《婴幼儿配方食品和乳粉通用检验方法(GB/T5413.30-1997)》乳、乳粉类杂质度测定的整体内容看,其前后内容矛盾,专有名词矛盾,技术内容不完整,甚至找不到所指的内容。这使原起草人都很难看懂。其前言中说只“对文本格式进行修改;内容未做改动”。但实际却增加了棉质过滤板的密度和一个附录A杂质度过滤板的检验。在棉质过滤板内容中,既然增加了密度就应该进一步明确它的材质、加工工艺、色泽、厚度、孔径、尘埃度、透气度等基本技术特征标准,没有明确这些特征技术标准,就会在执行这一检验标准时造成混乱。这也是市场上出现假冒伪劣的所谓“杂质度过滤板”的原因之一。在标准中同时出现了“杂质度过滤板”和“棉质过滤板”,而这两个不同的专有名词代表了两个不同的产品,而在同一个检验方法国家标准中同时出现,就会在执行该标准中造成混乱,这也是市场上出现假冒伪劣的所谓“杂质度过滤板”的第二个原因。杂质度过滤板有多个特征技术标准,不能用附录A中的方法检出,不知道附录A中的方法及内容要检验什么指标,而且附录 A中的某些标题找不到其内容,检验无法进行。从目前国家标准这一层面上讲,国家标准《婴幼儿配方食品和乳粉通用检验方法(GB/T5413.30-1997)应尽早修订为妥。
高纯金属中杂质元素(0.0001%量级含量)的含量测定是对分析中的一种挑战。而目前主要的分析方法有电感耦合等离子体质谱法(ICP-MS)和电感耦合等离子体发射光谱法(ICP-OES)等。本文就ICPMS分析八氧化三铀中杂质元素的方法进行介绍。1 前言核燃料铀是从天然含铀矿石中提取出来的,其间经过酸法(或碱法)浸出、湿法冶金、精制、同位素分离等一系列生产过程,最后制成核燃料元件或军用核产品。铀矿石中伴生着各种元素,而且从含铀矿物中提取铀到精制成核纯级铀需经过多次纯化富集,每经一个提取纯化过程,使产品纯度得到提高的同时,又由于每个工艺环节不同,加入试剂不纯而引入一些杂质。为了鉴定铀产品八氧化三铀的纯度,一般需要对产品中所含的各种微量杂质元素进行测定。目前较普遍的做法是将铀进行分离,利用分离之后得到的溶液进行杂质元素测量。但是经过树脂分离,增加了引入杂质元素的机会,使得分离过程中本底不容易控制,并且元素的回收率差别较大,对结果也有较大影响。而直接测定的方法基体较大,往往导致测量结果差别过大。本文讨论利用ICP-MS直接测定Cr、Mn、Ni、Cu、Mo等杂质元素,通过标准物质进行校正后,得到样品的测试结果,通过测试标准物质和实验室间比对认为ICPMS直接测定的方法是可行的。2 试验部分2.1 仪器和试剂NexION300D型电感耦合等离子体质谱仪,美国铂金埃尔默(PE)公司生产,仪器工作条件:发射功率1500W;等离子气流量:17.0L/min;辅助气流量:1.2L min;雾化气流量0.93 L min。精密电子天平:感量0.0001g,梅特勒-托利多仪器(上海)有限公司;自动控温电热板:常温至350℃,控制精密度±5℃。所用硝酸为MOS级;所有用水均为二次去离子水,电导率18.25MΩ/cm。标准溶液:混标溶液IV-ICP-MS71,浓度分别为10 ug/mL,稀释至0.5 ug/L、1 ug/L、5 ug/L、10ug/L;介质为1%的HNO3。2.2 试验方法实验步骤:称取0.05g八氧化三铀样品,加浓硝酸2mL放置在电热板上消解,蒸至近干后取下冷却至室温,加入1mL硝酸溶液样品完全溶解后移至100mL的容量瓶中进行定容。按照相同的方法制备GBW04242、GBW04243和 GBW04205的标准样品。同时按照上述步骤得到平行空白溶液。测量方法:使用10ug/L的Rh溶液为在线内标,通过一个Y形三通与样品溶液同时引入ICP仪器中进行测量。GBW04242、GBW04243当标准物质使用,而GBW04205当成未知样品进行方法验证。3 结果讨论3.1方法检出限在上述选定的样品前处理方法和仪器测试参数条件下,以样品平行空白连续测定11次,以3倍标准偏差计算方法检出限,见表1。表1 检测限 元素 分析质量数 空白浓度范围 ρ/(μg·L-1) 平均值 ρ/(μg·L-1) RSD /% 检出限 ρ/(μg·L-1) Cr 52 0.012~0.016 0.014 7.9 0.0035 Mn 55 0.011~0.027 0.023 9.1 0.0064 Ni 60 0.012~0.028 0.024 8.7 0.0049 Cu 65 0.007~0.032 0.029 7.9 0.0069 Mo 98 0.0005~0.0014 0.0012 9.7 0.0018 3.2加标回收和准确度将其按照实验条件将U3O8样品完全消解后稀释成10份溶液,取5份分别加入一定浓度和体积的单元素标准溶液,定容后上机测试溶液中元素浓度;另外5份加同体积的空白溶液做参照,来计算标准加入回收率,同时测定五次重测量的稳定性,结果显示Cr、Mn、Ni、Cu、Mo的回收率在86-112%之间。对国家标准物质GBW04205参考样品按照本方法实验条件进行分析测量进行计算,得到的结果与参考值接近。3.3样品测量结果对通过该方法测试得到如下结果。表2 样品测试结果 测量元素含量(μg/g)Cr24.5±1.8Mn13.1±1.1Ni44.5±3.8Cu18.1±1.6Mo8.35±1.24 ICP-MS直接测定法总结1 实验数据结果表明每个元素均可获得较低的检出限,回收率也在86%~112%之间,对照标准物质分析,结果表明本方法结果准确可靠。2 直接测定需要已经浓度的参考物质进行校正,如果没有合适的标准物质,不适合使用直接测定的方法。可采用基体匹配法,配制含有与样品相同基体的标准溶液进行分析测试。
哪位有高纯金杂质icp分析方法?
在做乙醇中挥发性杂质时,看过好多人的谱图,都说是杂质1,杂质2,我想问问杂质1,杂质2具体是什么物质?求解答。
如题,纯度在99%以上的纯金属,是测杂质含量然后用100%减去杂质来计算金属含量吗?有这方面的标准码?像纯铜和纯镍,如何定义杂质元素,需要测那些杂质元素?我记得好像有个纯铝含量的测定标准,就是这种差减法。
哪位有高纯铜杂质icp分析方法?
哪位有高纯铅杂质icp分析方法?
最近使用国内某地生产的硅胶过柱,样品一直含有邻苯二甲酸二异丁酯这个杂质。非常难以除去。问一下;国内有什么地方生产好的硅胶? 这个杂质一般怎么处理?是不是样品浓度大的话就可以掩盖这个杂质峰?
请问多晶硅行业三氯氢硅主要控制的金属杂质有哪些?控制指标是多少?大家一起讨论一下。我们这边是把混标中有的元素都测出来,但是具体控制的指标没有定论。







 我要推广仪器
我要推广仪器
 下载APP
下载APP