各位网友,我在做一个课题,关于氟氯苯菊酯的课题,现在的问题是所用仪器响应不是很好,现在我用的仪器是ECD和GCMS,征求各位网友。谢谢[em0810]
求助,有做农残[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]的老师吗?我们扩GB23200.121氟氰戊菊酯、氰戊菊酯、联苯菊酯、溴氰菊酯这四个菊酯,但是优化不出来。
各位文友,我正在做关于氟氯苯菊酯的课题,使用仪器有ECD和GCMS.但是响应不是很好,请问各位文友,有没有做过氟氯苯菊酯的?谢谢
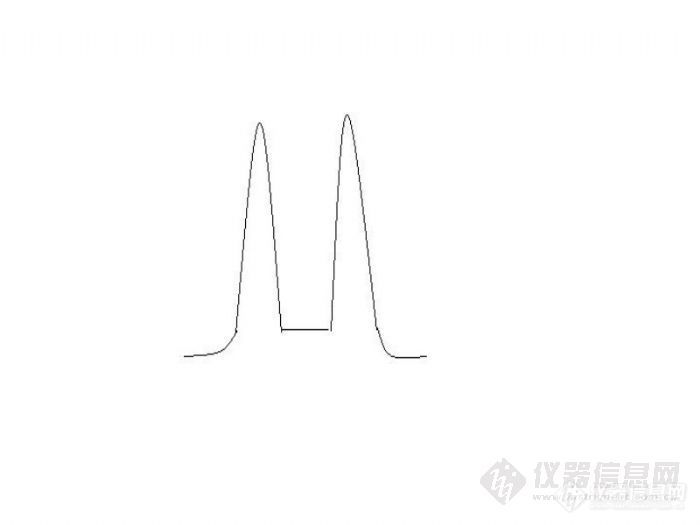
氯氰菊酯第四个峰和氟氰戊菊酯(氟氰菊酯)第一个峰无法分离,色谱柱CP-Sil 8 CB(0.25*0.25*30),使用zb-1701p和CP-Sil 19 CB(0.25*0.25*30),峰能分开,但各个异构体峰分离较差http://ng1.17img.cn/bbsfiles/images/2011/07/201107011433_302579_1830074_3.jpg
请教各位大侠:氢氟酸是否是强氧化性酸?能否与聚苯脂发生反应?聚苯脂就是聚对羟基苯甲酸苯脂??急用,在线等。我查了很多资料都没有相关的结论。
分享四份ISO标准:1. ISO 14389 : 2014 纺织品 -邻苯二甲酸酯含量的测定 -四氢呋喃方法2. ISO 13365-2011 皮革--化学试验--用液相色谱法测定皮革中防腐剂(TCMTB, PCMC, OPP, OIT)含量3. ISO 17070-2015 中文名称: 皮革--化学试验--四氯苯酚、三氯酚、二氯苯酚、氯酚-同分异构体和五氯苯酚含量的测定4. ISO 18254-1-2016 纺织品 烷基酚聚氧乙烯醚(APEO)的检测和测定方法.高效液相色谱-质谱法
请问有人知道氯烯炔、七氟甲醚、四氟甲醚菊酯液谱条件?正相的。谢谢各位高手啦。
蜂王浆样品用水溶解后经正已烷-二氯甲烷(1:1,V/V)提取,无水乙醇破除乳化,样液经离心后,用反相固相萃取柱富集与净化,用乙腈洗脱后,氮吹至近干,用80%乙腈水溶液溶解,经0.22μm滤膜过滤后,采用超高效液相色谱梯度洗脱与紫外检测器分析蜂王浆中的氟胺氰菊酯与氟氯苯氰菊酯残留。以XTerra Shield RP18柱(2.1mm×50mm,1.7μm)为色谱柱,以水和乙腈为流动相,两种菊酯残留在4min中内实现了较好的分离,方法快速准确,灵敏度高,是一种较好的定性和定量方法。所研究的提取净化方法及色谱分离条件能有效排除蜂王浆中的杂质干扰,添加回收率在75.7%~82.8%之间,方法检出限为10μg/kg。
你们这次有没有参加ACAS-PT478(2017)茶叶中联苯菊酯、毒死蜱的能力验证,有的话交流下实验哦
SNT 2320-2009 进出口食品中百菌清、苯氟磺胺、甲抑菌灵、克菌灵、灭菌丹、敌菌丹和四溴菊酯残留量的检测方法 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]质谱法
三氟氯氰菊酯的四种异构体必然同时存在么?
刚收到CNAS T0501茶叶中毒死蜱、联苯菊酯能力验证样品,有参加的朋友交流交流。
有参加福建出入境茶叶中联苯菊酯 毒死蜱能力验证的吗?交流一下 qq13724376
大家好,请问做农残检测时,有些组分会出现多个峰吗?比如我们做的三氟氯氰菊酯出现四个峰,像这种情况要怎么定量呢?谢谢!

[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱法测定毒死蜱、联苯菊酯不确定度评定1.编制目的:1.1给出茶叶中毒死蜱、联苯菊酯测定不确定度。1.2在测量结果处于临界状态时,用于对测量结果作出正确的判定。1.3用于评价实验室测量比对结果的质量。2.编制依据:2.1 JJF1059.1-2012《测量不确定度评定与表示》2.2 JJG 196-2006 《常用玻璃量器检定规程》2.3 CNAS-GL06:2006 《化学分析中不确定度的评估指南》2.5 GB/T23204-2008《茶叶中519种农药及相关化学品残留量的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱法》3.方法原理: 试样用乙腈提取,离心后,取上清液,经固相萃取柱净化,用丙酮+正己烷(1+1)洗脱后,氮吹定容后用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱仪检测。4.实验方法:4.1 标准溶液配制4.1.1. 毒死蜱、联苯菊酯标准储备液:准确称取毒死蜱0.02512g和联苯菊酯0.05024g,加丙酮溶解定容至50ml (V1),得到毒死蜱、联苯菊酯标准储备液浓度为(500+1000)μg/ml。4.1.2. 毒死蜱、联苯菊酯标准中间液配制毒死蜱、联苯菊酯标准中间液:用1ml移液管移取1.00ml (V2) 毒死蜱、联苯菊酯标准储备液(500+1000μg/ml)至50ml (V3) 容量瓶中,加丙酮+正己烷(1+1,v+v)定容至刻度,得到毒死蜱、联苯菊酯标准中间液的浓度为(10.0+20.0)μg/ml。 4.1.3. 毒死蜱、联苯菊酯标准上机液配制:用1ml移液管移取1.0ml (V4) 毒死蜱、联苯菊酯标准储备液(10.0+20.0μg/ml)至10ml (V5) 容量瓶中,加丙酮+正己烷(1+1,v+v)定容至刻度,得到毒死蜱、联苯菊酯标准中间液的浓度为(1.0+2.00)μg/ml。 4.2样品测定称样1.00g(精确到0.01g)样品于50mL离心管中,加入20mL乙腈,涡旋振荡3500r/min离心,取上清液乙腈20mL 于100mL鸡心瓶,40℃水浴旋蒸后洗脱转移至15ml离心管,氮吹至干,定容至1mL上机。5. 毒死蜱、联苯菊酯测量不确定度评定5.1 建立数学模型[img=,598,209]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011341570276_9155_2166779_3.png!w598x209.jpg[/img][img=,690,547]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011342035436_6301_2166779_3.png!w690x547.jpg[/img][img=,690,347]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011342117566_3961_2166779_3.png!w690x347.jpg[/img][img=,690,532]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011342193826_5254_2166779_3.png!w690x532.jpg[/img][img=,624,445]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011342257801_1661_2166779_3.png!w624x445.jpg[/img][img=,690,547]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011342318412_3679_2166779_3.png!w690x547.jpg[/img][img=,690,425]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011342400950_302_2166779_3.png!w690x425.jpg[/img][img=,690,580]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011342476327_1111_2166779_3.png!w690x580.jpg[/img][img=,537,419]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011342587594_240_2166779_3.png!w537x419.jpg[/img][img=,690,571]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011343087364_8661_2166779_3.png!w690x571.jpg[/img]即测量重复性带来的影响最大,因此应减小仪器重复性带来的误差,其次为茶叶称样质量m带来的影响,因此在检测时适当提高样品的称样量是有意义的。
分离甲氰菊酯、联苯菊酯、氟氰戊菊酯和氯氰菊酯用什么型号的色谱柱呢?现在用-1的色谱柱,联苯菊酯和甲氰菊酯分不开...求助各位前辈~
有奖问答’对错题: 涤纶是先从石油和炼焦副产品中提取原料聚合成聚苯甲酸乙二酯树脂,然后熔融纺丝成为涤纶!( )
有参加ACAS-PT304(2016)茶叶中联苯菊酯、毒死蜱能力验证的吗?加群交流一下啊 595456862
有参加中国检科院测试评价中心“ACAS-PT-304(2016)茶叶中联苯菊酯、毒死蜱测定能力验证“的朋友吗?可以站内联系;大家相互交流下。

茶叶中毒死蜱、联苯菊酯的测定(能力验证)今年9月中旬,我们收到了中国检验检疫科学研究测试中心举办的“茶叶中毒死蜱、联苯菊酯的测定”的能力验证2个独立样品。接到样品,我们就开始检测了。所用仪器:岛津[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]GC-2010plus(配ECD、FPD检测器);安捷伦7890B-5977A量品种效益年GCMS[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url]。样品前处理:参照GB/T23204-2008;称取2.00g的样品,同时加入20uL100ug/mL环氧七氯作为内标,加入20mL乙腈涡旋5min,于4000r/min转速下离心6min,吸取乙腈层的上清液于鸡心瓶中,再加入15mL乙腈重复提取一次,合并乙腈层提取液于鸡心瓶中减压旋蒸至近干,用cleanert TPT柱净化。净化后的茶叶提取液没有一点颜色,氮吹至近干,加入2mL正已烷溶解残渣。待测定。(小技巧)由于是参加能力验证,时间紧,茶叶的前处理又麻烦,一个提取液只有2mL,溶剂是正已烷,极易挥发,于是我们在进样瓶中放入[b]2ml色谱自动进样瓶内插管玻璃管[/b]作支管,将2mL的溶液分装成6个小瓶,这10个小瓶可独立作为检测,不污染及不损失,同时带一个有证书的质控样(证书附有毒死蜱、联苯菊酯的数值)[img=,690,1226]http://ng1.17img.cn/bbsfiles/images/2017/09/201709290822_01_2166779_3.jpg[/img]三个样品,这个场面壮观吧[img]http://simg.instrument.com.cn/bbs/images/default/em09502.gif[/img] 鉴于毒死蜱分子结构中也含有三个氯原子,先用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url](ECD)来检测毒死蜱、联苯菊酯,配一个单标(浓度均为1000ng/mL)来初测其大约含量。1000ng/mL混标色谱图(从图可以看出:毒死蜱在ECD上的响应信号也很好。)[img=,690,534]http://ng1.17img.cn/bbsfiles/images/2017/09/201709282003_01_2166779_3.png[/img]但是ECD基线波动比FPD更大,此时低含量的毒死蜱用ECD来检测就有很大的误差了。第二个样品的毒死蜱在ECD上的响应:[img=,672,537]http://ng1.17img.cn/bbsfiles/images/2017/09/201709282011_01_2166779_3.png[/img]从图中可以看出:毒死蜱检出很有可能是基线的一点小波动而被积分得到的,因此有可以造成误判。联苯菊酯自然是没有问题的。改用FPD作检测器,单独测定毒死蜱的含量。FPD检测毒死蜱的结果:第一个样品在FPD上的响应:[img=,690,434]http://ng1.17img.cn/bbsfiles/images/2017/09/201709282023_01_2166779_3.png[/img]第二个样品在FPD上的响应(此时为未检出了)[img=,690,483]http://ng1.17img.cn/bbsfiles/images/2017/09/201709282024_01_2166779_3.png[/img]带证书的质控样的测定结果[img=,679,495]http://ng1.17img.cn/bbsfiles/images/2017/09/201709282024_02_2166779_3.png[/img]严格按GB/T23204-2008找一个茶叶空白基质来配制标液,用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]法检测:茶叶空白质谱TIC图:(未检出毒死蜱、环氧七氯、联苯菊酯)[img=,690,484]http://ng1.17img.cn/bbsfiles/images/2017/09/201709282057_01_2166779_3.png[/img]工作曲线的绘制:[img=,690,317]http://ng1.17img.cn/bbsfiles/images/2017/09/201709282103_01_2166779_3.png[/img]毒死蜱50ng/mL+联苯菊酯100ng/mL由茶叶基质配制得来的TIC图:[img=,690,189]http://ng1.17img.cn/bbsfiles/images/2017/09/201709282105_01_2166779_3.png[/img]7号峰为毒死蜱:19.489min;8号峰为环氧七氯:20.600;16号峰为联苯菊酯:27.726min。FPD对毒死蜱的检测限:50ng/mL毒死蜱标液在FPD上的响应:5ng/mL毒死蜱标液在FPD上的响应:[img=,667,442]http://ng1.17img.cn/bbsfiles/images/2017/09/201709282136_01_2166779_3.png[/img]实际才5ng/mL,而仪器软件上却计算得出45ng/mL,且S/N才1.5,可是误差之大。5ng/mL的毒死蜱在GCMS上的响应:[img=,690,455]http://ng1.17img.cn/bbsfiles/images/2017/09/201709282133_01_2166779_3.png[/img][img=,666,476]http://ng1.17img.cn/bbsfiles/images/2017/09/201709282133_02_2166779_3.png[/img]以上是由购买标液(北京坛墨质检)配制得到的(这个作为方案一的工作曲线)。由于是能力验证,同时也为了验证各种不同厂家之间标准物质对测定的结果是否有差异及不同的配制方法对测定结果的影响,决定进行如下方案进行比对。方案二:由中国环境研究所购买的固标毒死蜱、联苯菊酯,用茶叶基质配制。方案三:不用茶叶空白基质,用正已烷来配制工作曲线。方案四:换另外一名同事来配制工作曲线。测定的结果如下:[img=,690,170]http://ng1.17img.cn/bbsfiles/images/2017/09/201709300949_01_2166779_3.png[/img]总结:1、用ECD虽然也可以检测毒死蜱,但是ECD的基线没有FPD平稳,固对于低含量的毒死蜱,用FPD检测器比ECD检测器可信度高。 2、用FPD检测毒死蜱的检出限为50ng/mL,而用GCMS检测毒死蜱的检测限可达5ng/mL,因此GCMS的灵敏度比FPD的高。 3、用茶叶空白基质与用正已烷配制的工作曲线,对检测结果的影响不大,用茶叶空白基质配制的比用正已烷配制的结果高10%左右,在可接受的范围之内。 4、用不同厂家之间的标准物质、不同的人员来配制工作使用标准溶液,以抵消人员,标准物质之间的误差 5、做能力验证,最好能带入与基质相同的带证书的质控样,这样比单纯做加标回收更可靠。如果质控样的数据与证书会对得上,这样会增大对样品测定结果的自信心。
求助苯醚菊酯中右旋苯醚菊酯的测定,谢谢
如何测定苯醚菊酯中右旋苯醚菊酯的含量,谢谢,461261985,qq
第一组,氯氰菊酯和高效氯氰菊酯,GB2300.8种查到氯氰菊酯的离子为181,152,180但是没找到高效氯氰菊酯,找到了一个顺式-氯氰菊酯离子为163,181,165.这个顺式-氯氰菊酯是高效氯氰菊酯么,但是英文名对不上啊,氯氰菊酯的四个峰是不是包括了高效的,求高手解答。第二组,氟氯氰菊酯和高效氟氯氰菊酯,23200.8只查到了,高效的181,197,141氟氯氰菊酯没有找到,氟氯氰菊酯又名三氟氯氰菊酯,能不能用高效的离子呢?
新人做[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url],四溴氟苯峰做内标时峰型正常,进样品时峰分叉,3个样品3根柱子,3个都是这样的情况,求助大神解疑答惑。
各位大虾:小弟最近忙着做农药残留(浓缩水果汁中的毒死蜱、马拉硫磷、溴氰菊酯、氯氟氰菊酯)的能力验证,但是实验过程中出现了问题,不知道错在哪里请大家帮助,我按照NY/T761-2004方法作的,最后结果很吓人,马拉硫磷的回收率分别为360%和430%。试验过程如下:称5克样品加入1ug/mL的毒死蜱与马拉硫磷的混标3mL,最后毒死蜱的回收率分别为80%和94%,但是马拉硫磷的回收率分别为360%和430%,而我加入的是混标不可能两种农药加标出错,请问这是什麽原因?请大家帮助我找找原因,只要回帖就送分!!很着急,5号上报结果啊!!
氯氟氢菊酯出了两个峰是分别积分还是积一起。积一起算一个结果怎么算的
请问有没有哪位老师做过纺织品中氟硅菊酯(分子式:C25-H29-F-O2-Si),根据国标GB/T 18412.4-2006,我用ECD检测器检测,1ppm在柱子上也没有响应,而在GC-MSD上有很好的响应,这是为什么啊?
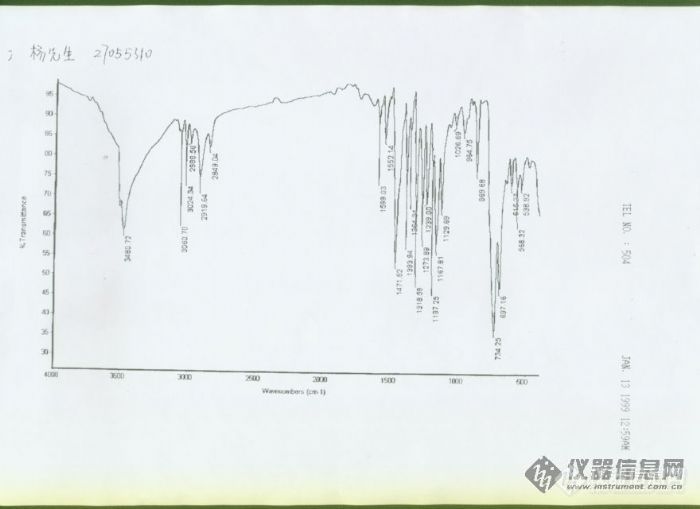
我用聚苯乙烯和四溴双酚A制成这个产品,我对照了一下聚苯乙烯、四溴双酚A的红外图谱,两张图合在一起与我产品的图谱差不多,但有一些吸收带出现在了变化,小弟对这些变化看不出所以然。大概主要是OH键。请各位帮我分析一下,此外,请问深圳有没有专门的测试机构?谢谢[img]http://ng1.17img.cn/bbsfiles/images/2006/01/200601191122_13291_1332078_3.jpg[/img]
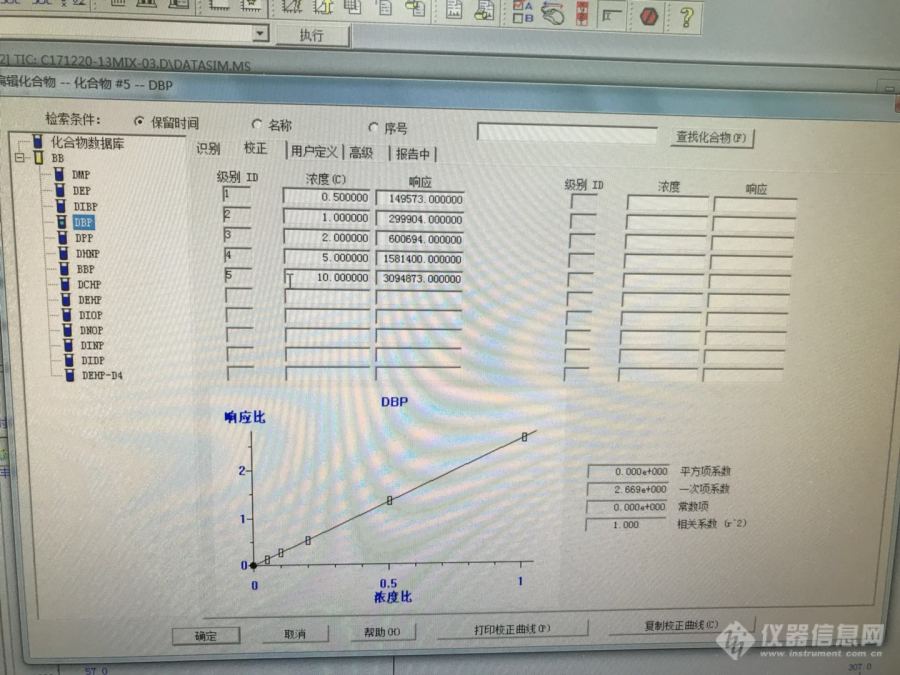
最近,做邻苯二甲酸酯的曲线,虽然线性有0.995以上,但是DEHP之后出峰的物质(DIOP,DNOP,DINP,DIDP),出现较大的负截距,对结果影响还是挺大的。而且是同一套曲线标液,有时后走出来又能ok,没问题。所以可以排除是人为配置的问题。配置曲线的基体空白也是背景不高。仪器的安捷伦的7890A/5975C.首先我是排查是否漏气,没问题,顺便也换了新衬管,结果还是没改善。今天我试着截掉色谱柱,也洗了分流平板,期待明天的结果。想问下,论坛的专家有没有其它可以参考的指导意见[img]http://simg.instrument.com.cn/bbs/images/default/em09502.gif[/img]下那面是两张图[img=1,533,400]http://ng1.17img.cn/bbsfiles/images/2017/12/201712222306_8863_2978213_3.jpg!w690x517.jpg[/img][img=2,533,400]http://ng1.17img.cn/bbsfiles/images/2017/12/201712222305_510_2978213_3.jpg!w690x517.jpg[/img]
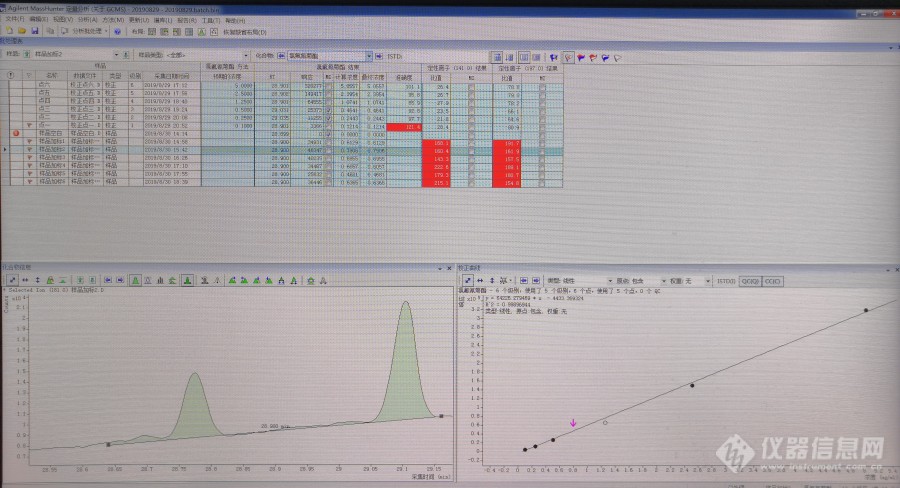
安捷伦[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]做嘧霉胺,氟虫腈,虫螨腈,甲拌磷砜,啶虫脒,氯氟氰菊酯,吡菌磷,哒螨灵,苯醚甲环唑的时候碰到的若干问题。仪器条件如下进样量:1ul。进样口温度:290℃。进样方式:无分流进样,1.5min后打开分流阀和隔垫吹扫阀。色谱柱型号:HP-5。载气:氦气,纯度≥99.999%,流速1.2ml/min。电子轰击源:70eV。离子源温度:230℃。GC-MS接口温度:280度。选择例子监测:每种化合物分别选择一个定量离子,2个定性离子。色谱柱温度程序:40℃保持1min,然后以30℃/min程序升温至130℃,再以5℃/min升温至250℃,再以10℃/min升温至300℃,保持5min。问题一,氯氟氰菊酯的定性离子。做添加回收的时候,标准曲线的比值和添加样品的比值相差很大,标准的定性离子的比值大约是26和75左右,添加样品就去到160和180左右。想知道哪里出现了问题,或者加入没有操作错误的话,怎么处理。[img=氯氟氰菊酯,690,374]https://ng1.17img.cn/bbsfiles/images/2019/09/201909021112439548_8430_2342324_3.jpg!w690x374.jpg[/img]问题二,甲拌磷砜,跑样品空白的时候,定量离子有检出,但是定性离子比值匹配,我就认为这个峰不是甲拌磷砜,删除了。但是在分析加标样品的时候,基本回收率都超高,我怀疑样品空白的那个定量离子和我的加标峰叠在一起了 这个怎么处理。







 我要推广仪器
我要推广仪器
 下载APP
下载APP