
在甲基培尼皮质醇的制造过程中,可能会生成一些杂质。这些杂质可能会出现在原料中,也可能在制药过程中的化学反应中产生。不论其来源,杂质的存在都可能影响到药物的质量、安全性和疗效。例如,甲基培尼皮质杂质可能增加药物的毒性,或导致不良反应。同样,杂质也可能对甲基培尼皮质醇的药效产生影响。因此,制药公司必须在生产过程中严格检测和控制这些杂质。检测和控制药品中的杂质是药品质量控制的重要组成部分。CATO标准品对杂质的研究不仅有助于保证药品的质量和安全性,也可以为优化制药过程提供参考。比如,通过对杂质的研究,可以找到产生这些杂质的原因,从而改进制药过程,减少杂质的生成。[img=,600,588]https://ng1.17img.cn/bbsfiles/images/2024/02/202402052058222428_1748_6381668_3.png!w600x588.jpg[/img]
间羟胺是一种有机化合物,主要用作还原剂和抗氧化剂,也用于一些特殊的有机合成反应。然而,它也是某些药物和化妆品制造过程中的杂质,如果含量过高,可能对人体健康产生不利影响。间羟胺杂质如果未经妥善处理,可能会导致药物或化妆品的质量下降,甚至影响药效。一些间羟胺化合物对人体有毒,长期或大量接触可能会引起皮肤和眼睛的刺激,甚至影响肝脏和肾脏,或引发神经系统和呼吸道问题。除此之外,间羟胺对环境也有潜在危害,过量的间羟胺可能污染水源,对水生生物和环境产生破坏。CATO标准品格外关注间羟胺杂质的存在是十分重要的,需要通过科学严谨的检测和控制技术,保证药物和化妆品的质量和安全。[img=,597,562]https://ng1.17img.cn/bbsfiles/images/2024/02/202402052113508399_5763_6381668_3.png!w597x562.jpg[/img]
大家,谁用ICP做过NMP(N-甲基吡咯烷酮)中杂质含量分析啊,怎么处理样品啊,大家积极讨论啊
做乙醇挥发性杂质,按药典做供试品b的4-甲基-2戊醇峰面积始终小于杂质峰,新人求指导
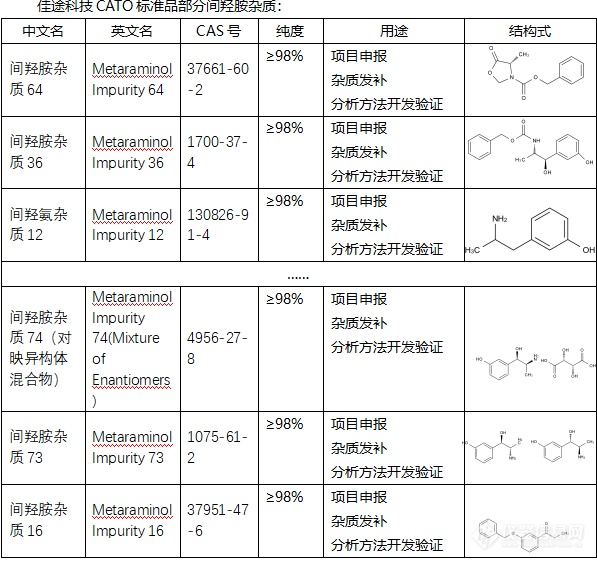
[align=center][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法分析甲基丙烯酸甲酯纯度及杂质的测定[/align][align=center]袁白杨[/align][align=center](宁波科元精化有限公司)[/align][align=left][color=black]摘要[/color][color=black]:建立了采用聚乙二醇色谱柱分析甲基丙烯甲酯纯度及杂质的测定方法。该方法采用氢火焰离子检测器(FID)进行分析,采用外标法进行定量。该方法的特点是操作简便、快速、样品用量少,结果准确,对于杂质的分析更全面。[/color][color=black]关键词[/color][color=black]:[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],聚乙二醇,甲基丙烯酸甲酯[/color]前言甲基丙烯酸甲酯(MMA)是一种重要的有机化工原料,主要用于生产聚甲基丙烯酸甲酯(有机玻璃)、聚氯乙烯助剂ARC和用作腈纶生产的第二单体,也可与其他乙烯基单体共聚得到不同性质的产品用作树脂、胶粘剂、涂料、离子交换树脂、纸张上光剂、纺织印染助剂、皮革处理剂、润滑油添加剂、原油降凝剂、木材和软木材的侵润剂、电机线圈的浸透剂、绝缘灌注材料和塑料型乳液的增塑剂等,用途十分广泛。甲基丙烯酸甲酯作为重要的化工原料,其杂质含量有严格的要求。目前普遍采用的是HP-5的毛细管柱,通过实验发现使用HP-5色谱柱时,各组分拖尾较为严重,且由于丙酸乙酯与甲基丙烯酸甲酯沸点接近,分析时无法分离,而使用聚乙二醇毛细管柱不仅各组分出峰对称,而且各组分均能完全分离。1 实验部分1.1 试剂 [size=16px]标准样品:含甲酸甲酯、丙酮、乙酸甲酯、甲醇、丙酸甲酯、异丁酸甲酯、丙烯酸甲酯、丙酸乙酯、甲基丙烯酸甲酯、甲基丙烯酸异丙酯、甲基丙烯酸乙酯、α-羟基异丁酸甲酯12种组分。[/size]1.2 仪器1.2.1 色谱仪:磐诺A91[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]1.2.2 检测器:FID检测器1.2.3 色谱柱:聚乙二醇毛细管柱1.3 色谱条件1.3.1 柱箱:初温:60℃恒5min,以30℃/min升至200℃,恒0.33min(总计10min);1.3.2 检测器:温度250℃, 燃气H2:35ml/min,助燃气Air:350ml/min;1.3.3 载气:N2,模式:恒压,线速度24cm/sec,分流比;30:1,仪器平衡时间1min。2 结果与讨论2.1 色谱图中组分的定性在相同实验条件下利用标准物质对各组分进行分析,采用保留时间定性,用标准物质混合样分析结果如图1。各组分都能在色谱图上出峰,且峰形对称,能够完全分离。[img]https://ng1.17img.cn/bbsfiles/images/2020/06/202006011345151848_1682_3989257_3.png[/img][/align][align=center]图 1[/align]而根据HG∕T 2305-2017方法中使用HP-5色谱柱进行分析时,发现丙酸乙酯与甲基丙烯酸甲酯完全重合,无法分离,如图2。从图中发现使用HP-5分析时,各组分拖尾较为严重。[img]https://ng1.17img.cn/bbsfiles/images/2020/06/202006011345159377_5083_3989257_3.png[/img][align=center]图 2[/align]2.2 定量分析采用外标法测定杂质组分的含量。计算方法根据标样浓度和峰面积的一一对应关系,求得样品中各组分的含量。[img]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181703262732_9921_3989257_3.png[/img][color=black]式中:[/color][color=black]—样品中某杂质的含量,%(m/m);[/color][color=black]—样品中某杂质的峰面积;[/color][color=black]—标样中杂质的含量,%(m/m);[/color][color=black]—标样中杂质的峰面积。[/color]2.3 精密度和检测限测定通过相同色谱条件下连续测定标准液体中各杂质的含量,来观察方法、仪器的精密度。数据见表1。根据2.5~3倍信噪比,可计算出检测限约为0.001%。[table][tr][td][align=center]组分[/align][/td][td][align=center]平均值(%)[/align][/td][td][align=center]标准偏差(n=6)[/align][/td][td][align=center]相对标准偏差/%[/align][/td][/tr][tr][td][align=center][color=black]甲酸甲酯[/color][/align][/td][td][align=center][color=black]0.164[/color][/align][/td][td][align=center]0.41[/align][/td][td][align=center]2.5[/align][/td][/tr][tr][td][align=center][color=black]丙酮[/color][/align][/td][td][align=center][color=black]0.164[/color][/align][/td][td][align=center]0.13[/align][/td][td][align=center]1.9[/align][/td][/tr][tr][td][align=center][color=black]乙酸甲酯[/color][/align][/td][td][align=center][color=black]0.333[/color][/align][/td][td][align=center]0.05[/align][/td][td][align=center]2.3[/align][/td][/tr][tr][td][align=center][color=black]甲醇[/color][/align][/td][td][align=center][color=black]0.169[/color][/align][/td][td][align=center]0.32[/align][/td][td][align=center]2.8[/align][/td][/tr][tr][td][align=center][color=black]异丁酸甲酯[/color][/align][/td][td][align=center][color=black]0.083[/color][/align][/td][td][align=center]0.06[/align][/td][td][align=center]1.6[/align][/td][/tr][tr][td][align=center][color=black]丙烯酸甲酯[/color][/align][/td][td][align=center][color=black]0.319[/color][/align][/td][td][align=center]0.30[/align][/td][td][align=center]2.9[/align][/td][/tr][tr][td][align=center][color=black]丙酸甲酯[/color][/align][/td][td][align=center][color=black]0.082[/color][/align][/td][td][align=center]0.01[/align][/td][td][align=center]1.8[/align][/td][/tr][tr][td][align=center][color=black]丙酸乙酯[/color][/align][/td][td][align=center][color=black]0.085[/color][/align][/td][td][align=center]0.22[/align][/td][td][align=center]2.0[/align][/td][/tr][tr][td][align=center][color=black]甲基丙烯酸异[/color][/align][align=center][color=black]丙酯[/color][/align][/td][td][align=center][color=black]0.363[/color][/align][/td][td][align=center]0.15[/align][/td][td][align=center]1.5[/align][/td][/tr][tr][td][align=center][color=black]甲基丙烯酸乙酯[/color][/align][/td][td][align=center][color=black]0.315[/color][/align][/td][td][align=center]0.18[/align][/td][td][align=center]2.4[/align][/td][/tr][tr][td][align=center][color=black]α[/color][font=宋体][color=black]-羟基异丁酸甲酯[/color][/font][/align][/td][td][align=center][color=black]0.175[/color][/align][/td][td][align=center]0.15[/align][/td][td][align=center]1.9[/align][/td][/tr][/table][align=center]表 1[/align]3 结论利用聚乙二醇色谱柱,全面测定了甲基丙烯酸甲酯中杂质的含量。[font=宋体][color=black]操作简便,[/color][/font][color=black]分离效果好,峰形对称,[/color][font=宋体][color=black]而且分析速度快[/color][/font][color=black],[/color][font=宋体][color=black]准确度高,为生产装置精细操作、[/color][/font][color=black]化工原料[/color][font=宋体][color=black]进一步深加工提供可靠的分析数据[/color][/font][size=13px]。[/size]参考文献吴烈钧. [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测方法. 化学工业出版社,1999汪正范. 色谱定性与定量. 化学工业出版社,2000HG/T 2305-2017 工业用甲基丙烯酸甲酯,2017杨海鹰. [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]在石油化工中的应用. 化学工业出版社,2004
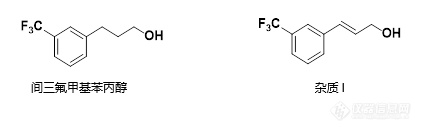
[align=center][b]间三氟甲基苯丙醇和杂质I的分离[/b][/align]客户提供了间三氟甲基苯丙醇和相关杂质I,并反馈曾尝试使用反相C[sub]18[/sub]柱对两化合物进行分离,但未能得到基线分离结果。现客户希望本实验室选择合适色谱柱并对色谱条件进行优化,来实现间氟甲基苯丙醇和其相关杂质I的基线分离。首先,我们尝试使用中等极性的CAPCELLPAK C[sub]18[/sub] MGII色谱柱,在磷酸盐-乙腈体系中分析50 μg/mL的混标溶液及各单标溶液,通过调整流动相中水相和有机相比例为60:40时,50 μg/mL的混标溶液中,间三氟甲基苯丙醇和杂质I能实现基线分离,分离度为1.52(见图1)。同客户沟通,客户希望供试品溶液(当间三氟甲基苯丙醇浓度为1mg/mL,杂质I为1 μg/mL)中两化合物分离度大于1.50。[align=center][img=,422,132]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009027392_4941_2222981_3.png!w422x132.jpg[/img][/align][align=center][img=,656,427]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009243004_918_2222981_3.png!w656x427.jpg[/img][/align][align=center]图1 MGII分析混标及单标溶液结果[/align][align=left][img=,575,197]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009245664_7431_2222981_3.png!w575x197.jpg[/img][/align][align=left]在此实验基础上,进一步分析供试品溶液,结果发现由于间三氟甲基苯丙醇浓度过高,致使色谱峰展宽,杂质I与间三氟甲基苯丙醇的分离度下降,未能达到1.50的基线分离要求;进一步尝试通过升高柱温来改善分离度,结果如图2,在50°C时能够得到良好分离结果,分离度为1.59。[/align][align=left][/align][align=center][img=,650,418]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031030364182_5088_2222981_3.png!w650x418.jpg[/img][/align][align=center]图2 MGII分析混标及单标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,575,195]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031031319132_5141_2222981_3.png!w575x195.jpg[/img][/align][align=left][/align][align=left]为有更多的选择,我们也尝试了两款非C[sub]18[/sub]色谱柱,包括键合特殊官能团——金刚烷基的高极性色谱柱ADME和键合五氟苯基的PFP色谱柱。在使用PFP色谱柱分析50 μg/mL混标溶液时,发现两化合物峰重合,未能实现分离。但使用ADME分析混标溶液时,能够得到1.36的分离度(见图3)。[/align][align=left][/align][align=center][img=,620,423]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034384978_3594_2222981_3.png!w620x423.jpg[/img][/align][align=center]图3 PFP、ADME分析50 μg/mL混标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,552,214]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034366042_2199_2222981_3.png!w552x214.jpg[/img][/align][align=left][/align][align=left]尝试改善分离度,继续使用ADME色谱柱进行分析,通过降低有机相比例来延长保留,最终得到了1.50的分离度(见图4),与此同时对供试品溶液进行分析,发现由于主成分峰展宽未能得到基线分离结果(见图5)。[/align][align=left][/align][align=center][img=,658,430]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035399180_5905_2222981_3.png!w658x430.jpg[/img][/align][align=center]图4 ADME分析混标溶液结果[/align][align=center][/align][align=center][img=,657,435]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035148034_8911_2222981_3.png!w657x435.jpg[/img][/align][align=center]图5 ADME分析供试品溶液结果[/align]注: 峰上标数字为分离度。[align=left][img=,586,223]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035150115_8050_2222981_3.png!w586x223.jpg[/img][/align]
请教各位高手,我换了预柱后,出现新杂质?请分析!波长:215nm,柱温40,柱子:Shodex RSpak DE-613.流动相为:0.6g 3-羟甲基-氨基甲烷,0.82mL辛胺到900mL水中,用0.3M盐酸调pH至2.5,用水定容到1000mL。样品溶剂就是流动相,但是换了预柱后,5min出现新的杂质。还有就是空白溶剂峰中有杂质?4min和9min左右,为啥?[img]http://ng1.17img.cn/bbsfiles/images/2009/10/200910312207_179416_1632673_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/10/200910312207_179417_1632673_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/10/200910312213_179419_1632673_3.jpg[/img]
各位老师好,我现在遇到一个方法,里面采用三羟甲基氨基甲烷作为缓冲液,但是待测物本身是没有酸碱性的,其杂质也没有酸碱性,为什么还要用缓冲液作为流动相的水相,也还是这个三羟甲基氨基甲烷有其他的用途?
SHT 1550-2000工业用甲基叔丁基醚(MTBE)纯度及烃类杂质的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法
5-羟甲基糠醛的检查方法及限度要求初探 摘要:5-羟甲基糠醛是含葡萄糖等单糖的注射剂中重要的有关物质。本文就5-羟甲基糠醛的来源、影响其产生的因素、检测方法及限度等进行了阐述。并提请申请人注意在制剂工艺筛选过程、以及质量研究过程中注意控制该杂质。关键词:5-羟甲基糠醛 检查方法 限度 一、概述 5-羟甲基糠醛(5-Hydroxymethyl furfural,简称5-HMF)是葡萄糖等单糖化合物在高温或弱酸等条件下脱水产生的一个醛类化合物,该化合物稳定性不好,容易分解成乙酰丙酸和甲酸,或发生聚合反应 摘自: 医 学教 育网www.med66.com 。一方面,5-HMF对人体横纹肌和内脏有损害,另一方面,葡萄糖注射液在高温情况下颜色容易变黄,虽然5-HMF本身无色,但由于5-HMF可发生聚合而由聚合物导致变色,且葡萄糖注射液颜色的深浅与5-HMF产生的量成正比,因此5-HMF的量可指代产品中葡萄糖的分解程度。由于5-HMF的毒性及对产品质量情况的指示作用,在含葡萄糖或其它单糖的制剂中应作为一个重要的有关物质加以控制。 影响5-HMF产生量的因素有溶液的pH值、灭菌的温度及灭菌的时间等。葡萄糖在碱性溶液中极不稳定,易脱水分解,而在酸性溶液中相对稳定,其中以pH3.0时分解最少,中国药典规定葡萄糖注射液的pH值应在3.2~5.5。葡萄糖注射液在高温加热灭菌时易产生5-HMF,其量的增加与灭菌温度、时间成正比,在工艺筛选过程中选择考察指标时,需注意纳入5-HMF检查项。另外在贮藏过程中,5-HMF也会增加,所以应尽量缩短葡萄糖注射液的贮藏时间。此外,有文献报道,不同方法制备的注射用水配制的葡萄糖注射液经灭菌处理后,5-HMF的量有显著性差异,四级截留法制备的注射用水较重蒸馏法、蒸馏法制备的注射用水更好。另据文献报道,亚硫酸盐在溶液中能与葡萄糖生成葡萄糖羟基亚硫酸,使葡萄糖分解成5-HMF的反应延迟。 医学教 育网收集整理 二、检查方法 5-羟甲基糠醛的检查方法通常有以下几种:控制溶液的颜色、紫外法、双波长法、杂质对照法等。 由于葡萄糖注射液颜色的深浅与5-HMF产生的量成正比,可通过控制溶液的颜色来控制5-HMF的量,但由于5-HMF并非葡萄糖注射液变色的唯一因素,该法专属性和准确性较差。 5-HMF的最大吸收波长为284nm,该处干扰较少,UV法常可以作为5-HMF检查的首选方法。如中国药典2000、BP2000、USP27和日本药局方中纳入的葡萄糖注射液,均采用UV法控制5-羟甲基糠醛含量。如果制剂中的主药或辅料在284nm有明显的吸收,会干扰5-HMF的检查,可采用HPLC法代替。 双波长法是通过试验找到与干扰组分在284nm波长处吸收度相同的等吸收波长,测定样品在这两个波长处吸收度的差值,以消除制剂中其它组分的干扰。如甲硝唑葡萄糖注射液,因甲硝唑在320nm 有最大吸收,在284nm、355nm 为等吸收波长,5%葡萄糖溶液在此两波长附近均无吸收,5-HMF在355nm 无吸收。因此,有生产单位选择284nm为测定波长,355nm 为参比波长,以两波长的吸收度差值(ΔA = A284 - A355) 为定量信息测定5-HMF的含量。需要注意的是,等吸收波长的准确性直接影响测定结果,因此需要进行完备的方法学验证,一般建议采用多个浓度进行试验。 采用HPLC-杂质对照法检查5-HMF,具有较强的专属性,一般采用的色谱条件为:ODS柱,甲醇-水作流动相,也可加入适量的酸调节pH值,在284nm波长下进行限度检查。在研究过程中应注意进行方法学考察,确定5-HMF的色谱峰不受其它峰的干扰。 各国药典对葡萄糖注射液中5-HMF的量进行了控制,方法均采用测定284nm处的吸收度,但对供试液的浓度和限度的规定有所不同,见表1。 以葡萄糖注射液为载体的输液剂应在质量标准中控制5-HMF;含葡萄糖等单糖的口服制剂,在制备工艺中有酸性环境或要经过高温过程的,一般也应对5-HMF进行控制。在制订5-HMF的限度时应注意:若是采用紫外法,包括双波长法,限度可参照中国药典中葡萄糖注射液或氯化钠注射液的相关规定;若采用HPLC法,根据文献报道的数据及申请人提供的研究数据,目前一般要求将5-HMF控制在0.025%(按葡萄糖含量计算)左右。 以上是作者在查阅文献资料的基础上,并结合审评的实际情况对5-羟甲基糠醛的简要小结,希望与各位同仁探讨交流。
很多物质在甲醇和乙腈中不能非常好的溶解,然后也容易附着在柱子上,比如联苯类,44-二溴联苯,疏水性很强。但是这些杂质都能在强溶剂,比如四氢呋喃,二氯甲烷,DMSO等里面很好的溶解。所以我想问,能否使用一定比例的强溶剂清洗色谱柱。我打算用20%甲醇冲洗1h,在用100%甲醇冲洗1h,再100%乙腈1小时,同时进样一针20ul的四氢呋喃——再用甲醇:四氢呋喃50:50的流动相冲洗1h,进样一针20ul的DMSO——再甲醇:四氢呋喃50:50不进样DMSO 冲洗1h——再转回20%甲醇一小时,100%甲醇一小时,乙腈2h,最后保存在这个条件下。请问普通的C18柱能否用以上条件冲洗?如不可以,是否有更好的方式来清洗强杂质,再生色谱柱望指教。
供试品溶液(b)中其他各杂质峰面积的总和不得大于4-甲基-2-戊醇的峰面积(0.03%,以4-甲基-2-戊醇计)。这个其他各杂质峰包不包括甲醇、乙醛、乙缩醛和苯?求指点!
某原料药中,两个杂质的结构很相近,只差一个甲基,液相上两者的出峰位置也在同一时间,标准规定两个杂质和的限度为不超过0.5%,没有规定每一个单独杂质的限度,请问这两个杂质的方法学该怎样来做?要分开来做还是和在一块做,还是就单做其中的一个就行?
各位:以下是安宫黄体酮的杂质F 的TLC检测.可是按以下方法我做的薄层效果不好.不能分离,是何原因啊?用薄层色谱法检测 ,使用TLC硅胶色谱板。分别取加10μl的溶液点板,用10体积四氢呋喃试剂、45体积的1,1-二甲基乙基 甲基醚试剂和45体积的环己烷试剂配制的展开剂展开超过10cm,让色谱板在空气中晾干,然后用相同的展开剂在同一方向上再次展开超过10cm,加热色谱板到120℃并保持10min,用200g/l的甲苯亚璜酸乙醇溶液来喷板,加热到120℃并保持10min, 冷却。 在365nm的紫外光下检测,供试液(a)得到的色谱图中任一蓝色荧光斑点的Rf值比安宫黄体酮主斑点的Rf值要高,但是斑点的颜色不能深于参照液(a)得到色谱图中杂质F的蓝色荧光斑点(0.5%)。 只有当参照液(a)得到色谱图中有两个明显分离的斑点时,该测试结果才有效。
[font=&][size=16px][color=#333131]可以做氨丁三醇(三羟甲基)氨基甲烷,杂质峰定性的检测机构请看过来[url=https://www.woyaoce.cn/helptest/detail-dcb6af847ef4628458ecdf6d407180cd.html]点击查看详情[/url][/color][/size][/font]
常用辅料及相关杂质在系统一时保留时间参考值极性色谱柱 辅料或杂质名称 相对保留时间 异丙醇 0.098 正丙醇 0.168 二甲基甲酰胺 0.500 乳酸乙酯 0.512 二甲基乙酰胺 0.591 二甲基亚砜 0.785 1,2-丙二醇 0.803 1,3-丁二醇 0.956 1,3-丙二醇 1.000 苯甲醇 1.083 二甘醇 1.178 肉豆寇酸异丙酯 1.206 三乙酸甘油酯 1.263 丙三醇 1.469 油酸乙酯 1.564 苯甲酸苄酯 1.683 中等极性色谱柱辅料或杂质名称 相对保留时间乙醇 0.281 异丙醇 0.333 丙三醇 1.000 三甘醇 1.227 以上是从别的地方看到的,有的样品出峰时间非常接近。在新药申报时遇到的问题是:当同一个样品在做检出限时,很可能出峰时间出现较小的变化,会不会影响到样品种类判断及资料申报呢?
镀金层具有优良的抗变色、抗氧化和耐腐蚀性能 、良好 的芯片焊接和引线键合性能以及较低的接触电阻和较好的 可焊性等优点 ,被广泛应用于军用半导体及微电子封装外 壳。但军用电子器件对镀金层质量要求很高 ,而镀 金液中 的金属 杂质 则 直 接 影 响 镀 金 层 质 量 ,这 些 杂 质 主要 有 铅 、 铜 、铁 、镍等 ,往往在金结晶过程 中共沉积。其 中铅最有害 , 1~10mg/L就能造成非常有害的影响 ,特别是在低 电流密 度区 ;铜可使低电流密度区变暗,与金共沉积使颜色异常 , 纯度下降 ;铁 、镍等在酸性溶液或碱性亚硫酸盐槽液里 与金 共沉 积 的倾 向要 比在 碱 性氰 化 物 槽液 里 大得 多 ,对 金 的纯 度及颜色有害 。因此 ,准确测 定镀金液 中杂质元素 的含量 具有 重 要意 义 。目前 ,国内外对镀金液中杂质元素的测定虽有报道,但 多针对镀金液中单元素的分析研究 ,多元素的同时测定 多采 用 ICP—AES法 和 ICP—MS法 ,由 于镀 金 液 中大 量 基体元素金的存在对杂质元素测定的干扰和抑制作 用,高 盐样品直接进样导致进样系统堵塞和金的记忆效应等诸 多 问题,使得用 ICP—AES法直接测定镀金中杂质元素浓度相 当困难 。为此 ,研究 了用甲基异丁基酮(MIBK)有机试剂萃 取分离 了镀金液中的金后,采用 ICP—AES法测定镀金液中 Pb,cu,Fe,Ni4种杂质元素的方法 。为寻找镀金液中杂质元素的测定方法 ,运用甲基异丁基 酮(MIBK)萃取分离金 ,采用电感耦合等离子体原子发射光谱(ICP—AES)法对镀金液中的杂质元素进行了分析。对分析谱线、基体元素和等离子体参数等进行了讨论。结果表 明,这种方法的检出限为 0.008—0.019 g/mL,回收率为 89.4% 一102.3%,相对标准偏差 (RSD)小于 3.12% 。该法准确 、快速 、简便 ,应用于镀金 液 中杂质元素的测定 ,结果令人满意 。
[em09511]在葡萄糖注射液(包括常用的5%和10%两种)有效期内(一般以三年考察期内计),它的杂质5-HMF一般上升2倍左右,葡萄糖氯化钠注射液(含葡萄糖5%)在三年考察期内,它的杂质5-HMF量上升也在2倍左右。但如果葡萄糖与喹诺酮类的产品,如氟罗沙星配制成注射液,则在一年内变化值即可超过3倍,一般两年内A值均可以达到0.32(可用液相法进行折算)。国家还没有统一这类复方制剂的5-HMF检测方法,我们只是自己研究制订了氟罗沙星葡萄糖注射液中5-HMF检测方法,仅供参考:用十八烷基硅烷键合硅胶为填充剂;以三乙胺磷酸溶液(取三乙胺5ml和磷酸7ml,加水至1000ml)-乙腈(82:18)为流动相,检测波长为284nm,理论板数按5-羟甲基糠醛峰计算,应不低于2000。测定方法:精密量取供试品适量(约相当于葡萄糖1.0g),置100ml量瓶中,加流动相稀释至刻度,摇匀,做为供试液。另取5-HMF的对照品,加水溶解成每1ml约含2µ g的溶液作为对照品溶液。精密量取20µ l注入液相色谱仪,调整仪器灵敏度,使5-HMF峰面积为满量程的20%~25%。同法进行供试液的操作,记录色谱图,检查供试品溶液有无对照品溶液相同色谱峰。如有,供试品5-羟甲基糠醛按浓度( µ g/ml)表示。
做一个尿液中有机磷代谢物(二甲基磷酸酯,二乙基磷酸酯,二甲基硫代磷酸酯,二乙基硫代磷酸酯,二甲基二硫代磷酸酯,二乙基二硫代磷酸酯)的方法,检测物的极性非常强,在酸性条件下(PH1.25)的情况下,可用有机溶剂乙腈/乙醚提取,但是同时会提取出许多其它的极性杂质,比较复杂。弱问,有何去除杂质的方法?谢谢各位先
在粉末压片制样时,我们常常使用粘结剂,我常用的是甲基纤维素和淀粉,但里面都含有微量的金属杂质,不知道大家采用什么方法进行去除?用酸浸吗?希望大家献言献策。还有我们安装仪器前工程师还让我们准备了硬脂酸、硝酸锶,这两种物质是作什么用处啊
测定精对苯二甲酸(PTA)中的主要杂质对羧基苯甲醛(4-CBA)和对甲基苯甲酸(p-TOL)高效毛细管电泳仪法和高效液相色谱法,在测定精对苯二甲酸(PTA)中的主要杂质对羧基苯甲醛(4-CBA)和对甲基苯甲酸(p-TOL)时,哪种方法更好?听客户提到国标有用极普仪测试的,请问有没有对这个比较了解的。谢谢
就是我在做液相色谱分析的时候,分析3.5二甲基苯甲酸,在主峰出现之前出的杂质峰比以前大,这是请问这是什么原因?
怎么测试PVDF(聚偏氟乙烯/-(C2H2F2)n- )、CMC(羧甲基纤维素钠)、SBR(丁苯橡胶)杂质元素?
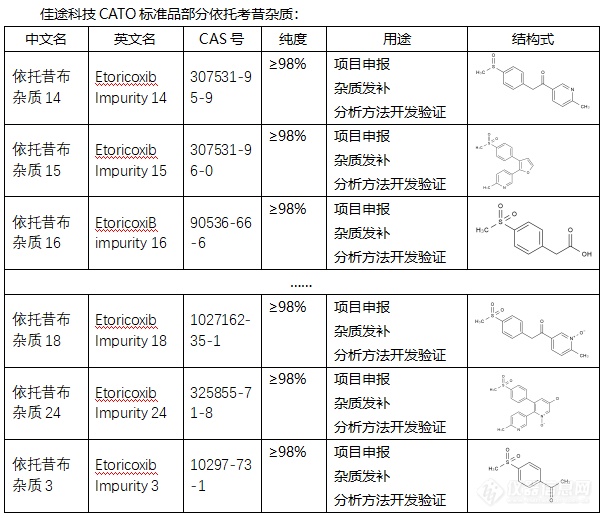
[font=宋体]◇依托考昔杂质[/font][font=宋体][font=宋体] 依托考昔杂质是在依托考昔的生产或保存过程中产生的非目标化合物。这些杂质可能会影响依托考昔的纯度和药效,因此在依托考昔的生产和质量控制过程中需要严格控制其含量。依托考昔杂质有多种类型,每一种都具有不同的化学特性,如[/font][font=Calibri]CAS[/font][font=宋体]号、分子式、分子量等。例如,依托考昔杂质[/font][font=Calibri]K[/font][font=宋体]的[/font][font=Calibri]CAS[/font][font=宋体]号为[/font][font=Calibri]349536-41-0[/font][font=宋体],分子式为[/font][font=Calibri]C18H15ClN2O3S[/font][font=宋体]。依托考昔杂质[/font][font=Calibri]Etoricoxib[/font][font=宋体]的[/font][font=Calibri]CAS[/font][font=宋体]号为[/font][font=Calibri]202409-33-4[/font][font=宋体],分子式为[/font][font=Calibri]C18H15N2O2SCl[/font][font=宋体],别名包括依托考昔、[/font][font=Calibri]5-[/font][font=宋体]氯[/font][font=Calibri]-2-(6-[/font][font=宋体]甲基吡啶[/font][font=Calibri]-3-[/font][font=宋体]基[/font][font=Calibri])-3-(4-[/font][font=宋体]甲基磺酰苯基[/font][font=Calibri])[/font][font=宋体]吡啶等。依托考昔杂质[/font][font=Calibri]Q[/font][font=宋体]的[/font][font=Calibri]CAS[/font][font=宋体]号为[/font][font=Calibri]292067-97-1[/font][font=宋体],分子式为[/font][font=Calibri]C18H15ClN2S[/font][font=宋体]。此外,依托考昔还可能存在其他未具体命名的杂质。[/font][/font][font=宋体][font=Calibri] CATO[/font][font=宋体]标准品提供的依托考昔全套的杂质[/font][/font][font=宋体],[/font][font=宋体]这些杂质对于药物的纯度和稳定性研究至关重要,也是药物研发过程中不可或缺的一部分[/font][font=宋体]。[img=,604,518]https://ng1.17img.cn/bbsfiles/images/2024/02/202402182043373327_8351_6381607_3.png!w604x518.jpg[/img][/font][font=宋体][color=#05073b][back=#fdfdfe] 广州[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]佳途科技[/back][/color][/font][font=宋体][color=#05073b][back=#fdfdfe]股份有限公司[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]深知药物研发与质量控制的重要性[/back][/color][/font][font=宋体][font=宋体],[/font][font=Calibri]CATO[/font][font=宋体]标准品厂家,提供依托考昔全套[/font][/font][font=宋体]的[/font][font=宋体]杂质,为客户提供更加精准、可靠的分析标准品,助力药物研发事业的快速发展[/font][font=宋体],[/font][font=宋体]以满足客户在药物研发和质量控制方面的需求。[/font]
请教大家,我的反应物进样GC-MS的时候显示有三个杂质,我的反应物是 4-chlorobiphenyl这三个杂质分别是 biphenyl ; 3-ethyl biphenyl; 4,4'-dichlorobiphenyl。为了不影响实验结果,我想想办法把这三个杂质去掉,尽量再提纯,虽然反应物买来的时候供应商说他们的纯度98.8%,但随着储存的时间长,貌似纯度越来越低了,以至于实验完了我没法判断我的产物是来自我的反应物还是这些杂质。请问大家有什么好方法来提纯我的反应物吗? 这几个物质结构相近,沸点也相差没有那么多。 虽然这个提纯的知识点高中就学了,但看了一遍竟然觉得没有一个可操作性强的……先行谢过各位大侠!!
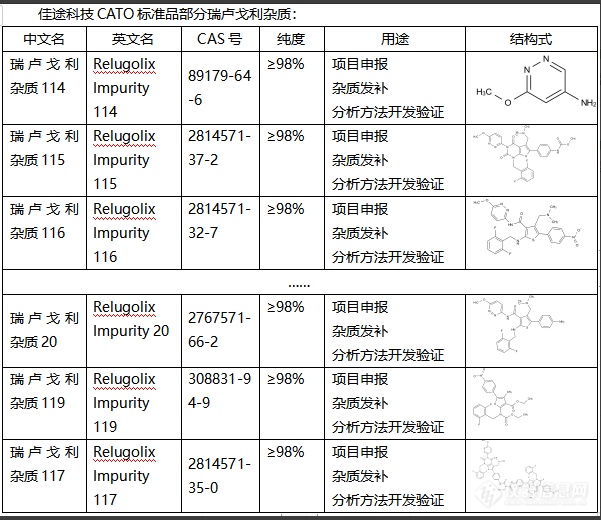
瑞卢戈利是一种硫酸盐矿物,也是重要的铁矿石。瑞卢戈利杂质对其性质、结构、颜色、光泽等都有重要影响。1.改变瑞卢戈利的颜色:瑞卢戈利杂质的存在可以改变瑞卢戈利的颜色,让它呈现出多种颜色变化,增加了瑞卢戈利的观赏性。2.影响瑞卢戈利的硬度:杂质的存在增强了瑞卢戈利的硬度,使其更能抗磨损,延长使用寿命。3.改变瑞卢戈利的比重:杂质的存在使瑞卢戈利的密度增大,从而改变了其比重,这对物理、化学等实验研究具有重要的参考价值。4.影响瑞卢戈利的光泽:杂质的存在可以增强瑞卢戈利的光泽,提高其观赏性。总的来说,瑞卢戈利杂质的作用是通过影响瑞卢戈利的物理和化学性质,达到改变其颜色、硬度、比重、光泽等目的,CATO标准品能对其在地质研究、工业生产等方面的利用提供帮助。[img=,601,520]https://ng1.17img.cn/bbsfiles/images/2024/02/202402021643477609_8603_6381668_3.png!w601x520.jpg[/img]
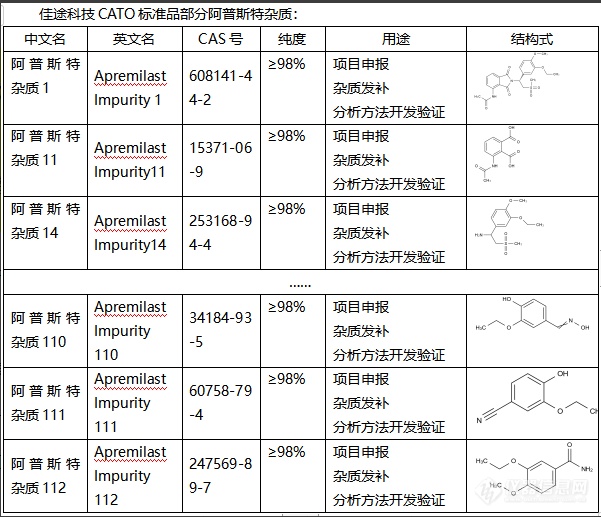
阿普斯特杂质(Acceptor impurities)在半导体中起到了非常关键的作用。1. 提供洞:阿普斯特杂质是电子受主,它会吸收自由电子,形成空穴(或称为“洞”)。这些空穴可以移动,起到电流传导的作用。因此,添加阿普斯特杂质后,半导体的导电性能会增强。2. 形成P型半导体:当阿普斯特杂质的浓度足够高时,半导体中的空穴数量将超过电子数量,形成了主导电流传导的是空穴的P型半导体。3. 局域能级:阿普斯特杂质也能产生在能带间的局域态,充当了能量级的“桥梁”,使电子更容易通过能阶间跃迁,也有助于电流的传导。CATO标准品改变半导体性质:通过改变阿普斯特杂质的种类和浓度,可以改变半导体的性质,如导电性、光学性质、磁性等,使之满足特定的使用需求。[img=,601,517]https://ng1.17img.cn/bbsfiles/images/2024/02/202402021700369834_2567_6381668_3.png!w601x517.jpg[/img]
2,6-二甲基氯苄做[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]时为什么后面出现个杂质酚,好像精制不掉的
请问从哪儿能查到海水中下面的杂质含量测定的方法:甲醇氯化石蜡-52乙二醇二乙二醇二甲基甲酰胺苯乙烯丙酮冰醋酸氯仿甲苯二甲苯1,4-丁二醇苯酚环氧乙烷
[B][center]药物中杂质的来源及杂质限量检查[/center] [/B]药物只有合格品与不合格品;一般化学试剂分为4个等级(基准试剂、优级纯、分析纯、化学纯) [B]药物中一般杂质检查 [/B][B]氯化物为一指示性杂质。[/B] 通过对氯化物的控制,可同时控制与氯化物结合的一些阳离子以及某些同时生成的副产物。可从氯化物检查结果显示药物的纯度,间接考核生产、贮藏过程是否正常。 1. 原理 药物中微量的氯化物在硝酸酸性条件下与硝酸银反应,生成氯化银的胶体微粒而显白色浑浊,与一定量的标准氯化钠溶液在相同条件下产生的氯化银浑浊程度比较,判定供试品中氯化物是否符合限量规定。 Ag+ + Cl- → AgCl ↓ [B]硫酸盐检查法 [/B] 1. 原理 药物中微量的硫酸盐在稀盐酸酸性条件下与氯化钡反应,生成硫酸钡的微粒而显白色浑浊,与一定量的标准硫酸钾溶液在相同条件下产生的硫酸钡浑浊程度比较,判定供试品中硫酸盐是否符合限量规定。 [B]铁盐检查法 [/B]硫氰酸盐法 巯基醋酸法 砷盐检查法 1. 古蔡氏法 1. 原理 金属锌与酸作用产生新生态的氢,与药物中微量砷盐反应生成具挥发性的砷化氢,遇溴化汞试纸产生黄色至棕色的砷斑,与同条件下一定量标准砷溶液所生成的砷比较斑,判断砷盐的含量。 [B]硒、氟及硫化物检查法 [/B]1. 氧瓶燃烧法 适用于以共价键结合的卤素、硫、硒的有机药物。 本法系将有机药物防入充满氧气的密闭燃烧瓶中进行燃烧,将燃烧所产生的欲测组分吸收于适当的吸收液中,然后根据欲测组分的性质,选用合适的分析方法进行鉴别、检查或含量测定。 [B]注意事项及讨论 [/B]1. 根据被燃烧分解的样品量选用适宜大小的燃烧瓶。 2. 测定氟化物时应改用石英燃烧瓶。 1. 硒检查法 (1). 操作方法 样品与对照品液,调节Ph2.0±0.2,加盐酸羟胺,二氨基萘,比色。 [B]硫化物检查法 [/B] 方法同砷盐检查第一法,不装醋酸铅棉花,以醋酸铅试纸代替溴化汞试纸。 标准液取1ml 5/ml [B]澄清度检查法 [/B]将一定浓度的供试品溶液与浊度标准液分别置于配对的比浊用玻璃管,同置黑色背景上,在漫射光下观察。浊度标准液 硫酸肼与乌洛托品溶液混合分五个等级,未超过0.5等级即为澄清。BP98规定未超过1等级即为澄清。 [B]溶液颜色检查法 [/B]CHP2000 [B]1. 比色法[/B] 色调标准贮备液 黄色液 重铬酸钾液(BP98用氯化铁) 红色液 氯化钴液 蓝色液 硫酸铜液 配成各种色调色号标准比色液共50种。 [B]2. 分光光度法 [/B] [B]易碳化物检查法 [/B]检查药物中含有的遇硫酸易碳化或易氧化而呈色的有机杂质。 对照品液 样品液 加硫酸5后,加供试品。 [B]炽灼残渣检查法[/B] 取供试品1.0~2.0g或个药品项下规定的重量,置已炽灼至恒重的坩埚中,精密称定,缓缓炽灼至完全碳化,放冷至室温;除另有规定外,加硫酸使湿润,低温加热至硫酸蒸气除尽后,在700~800炽灼使完全灰化,移至干燥器内,放冷至室温,精密称定,再在700~800炽灼至恒重,即得。残渣限量一般为0.1~0.2% 一般应使炽灼残渣量为1~2mg 若需将炽灼残渣留作重金属检查时,炽灼温度必须控制在500~600。 [B]干燥失重测定 [/B]1. 常压恒温干燥法 2. 干燥剂干燥法 3. 减压干燥法 [B]水分测定法 [/B][B]费休氏法 [/B] 本法是根据碘和二氧化硫在吡啶和甲醇溶液中能与水起定量反应的原理以测定水分。 [B]甲苯法[/B] 在加热状态下,甲苯夹带着水分蒸出,收集蒸出的水分测定。 [B]药物中特殊杂质检查 [/B] [B]一、物理法 [/B] [B]二、化学反应法 [/B](一)容量分析法 (二)重量分析法 (三)比色法和比浊法 [B]三、色谱法 [/B]1.纸色谱法 薄层色谱法 TLC是药典中最常用的特殊杂质限量检查方法。 1.在一定供试品及检查条件下,不允许有杂质斑点存在 2.以待测杂质对照品检测 3.将供试品稀释到适当浓度作为杂质对照品溶液 4.选用质量符合规定的与供试品相同的药物作为杂质对照品 [B]高效液相色谱法 [/B] [B][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法 [/B] 1.面积归一化法 2.主成分自身对照法 3.内标法测定 4.内标法加校正因子法 5.外标法 有机溶剂残留量测定法 [B]分光光度法 紫外分光光度法 比色法 [url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法[/B]







 我要推广仪器
我要推广仪器
 下载APP
下载APP