文献上用亚甲基蓝脱色力来衡量活性炭的能力,想问一下甲基蓝脱色力是不是就是标准上说的甲基蓝吸附量?如果不是,那它是什么?具体怎么算一直弄不明白谢谢了
急用亚甲基兰脱色方法请教亚甲基兰脱色方法的详细步骤,感激不尽,谢谢!!!
在做邻苯二甲酸盐时遇到的问题,用三氯甲烷:甲醇(2:1)索氏萃取PPO塑料中的邻苯二甲酸盐,得到的萃取液是比较深的蓝色(问了原来是这个塑料中添加了蓝色的色粉),想通层析柱去除。先用旋转蒸发仪蒸干,将溶剂置换为正己烷,接下来过层析。试了硅胶柱和氧化铝柱,结果都一样,用正己烷洗脱的话,得到的洗脱液是无色的,但是没有邻苯二甲酸酯;用三氯甲烷:正己烷(3:7)洗脱,洗脱液也是无色的,但是洗脱液中也没有邻苯二甲酸酯;只有用丙酮:正己烷(2:8)可以将邻苯二甲酸酯洗脱出来,但是由于这种洗脱液极性根强,把硅胶吸附的蓝色染料也洗脱下来了。不知道什么样的洗脱液比较合适?请问做层析的高手提供一个好方法,极性介于三氯甲烷:正己烷(3:7)和丙酮:正己烷(2:8)之间的洗脱液。
热脱附解吸率98%是怎么做出来的?问了几个色谱"大咖",他们说相同浓度和进样量,相同的色谱条件,液体进样和热脱附进样后对比峰面积。这么做 对吗?我们尝试做了,好像解吸率98%比较难做到?
热脱附解吸率98%是怎么做出来的?问了几个色谱"大咖",他们说相同浓度和进样量,相同的色谱条件,液体进样和热脱附进样后对比峰面积。这么做 对吗?我们尝试做了,好像解吸率98%比较难做到?
英蓝技术之三:英蓝稀释1、英蓝稀释的作用在样品浓度过高时,我们可以将样品稀释之后再通过超滤单元或者渗析单元,实际上,稀释的这个步骤也可以交给英蓝技术来完成。另一个方面,对于实测的数据变化过大的样品,我们也很难组织实验,例如,对于一批样品来说,其待测离子的浓度差相差巨大,数据点很可能并不落在标准曲线之上。如果通过人为的判断,然后再对每个样品进行不同倍数的稀释,会面临大量的人工劳动和值守判断,自动进样器将毫无用武之地。此外,如果是手工做稀释的话,经常需要用到容量瓶,而容量瓶的彻底清洗是非常困难的,稍有不慎就会造成污染,很多方案利用自动进样器上的蠕动泵来实现稀释,即向每个样品位加入一定量的溶剂;或者在自动进样器与色谱仪之间加入一个缓冲的单元,利用蠕动泵向缓冲单元中分别加入样品和溶剂,从而达到稀释的目的。这种通过蠕动泵完成的稀释常常精度不够,而且需要在一个自动进样器上集成多个蠕动泵并分别控制,管路复杂且难于清洗,因此限制了其应用的范围。英蓝稀释采用了完全不同的方法,英蓝稀释的加液过程并不是通过蠕动泵完成的,而是通过专门的Dosino加液单元来完成。Dosino加液单元可以看做是一个固定式、多通道、连续流、无交叉污染的精密配液器,其精度可以达到移液量的万分之一,正是有了Dosino的帮助,使得稀释的过程可以完全做到精确、无误。2、英蓝稀释的操作过程:1)Dosino加液单元分别将样品和溶剂按照比例加入到英蓝稀释单元中2)英蓝稀释单元对样品和溶剂进行充分的搅拌混合3)进样测量4)测量的同时进行逻辑判断,根据信号是否落在标准曲线的范围内判断理想的稀释的倍数并自动确定是否进行二次稀释或进样测量5)自动排空英蓝稀释单元并清洗3、英蓝稀释应用实例脱硫废水经英蓝稀释后检测淋洗液:NaHCO3/Na2CO3: 1.0/3.2 mM自动稀释100倍检测,20μl进样英蓝稀释实例1 Cl 1896 ppm2 NO3 362 ppm3 SO4 1341 ppmhttp://ng1.17img.cn/bbsfiles/images/2014/12/201412111117_526682_312_3.jpg
威玛龙色谱工作站里的梯度表“洗脱曲线”栏的内容意义是什么?
[color=#00008B][color=#DC143C]我们公司刚买的岛津lc020A的液相,我们专门打一个产品。用的流动相是 乙腈:0.1%磷酸水溶液=15:85色谱柱为 shim vp ods C-18柱仪器和色谱柱都是新买的,用了不到20次(大约买了才45天)前几次做样,出峰是正常的。昨天下午我分析产品也是正常的,用的是纯甲醇冲洗柱子,并保留的。等我今天下午在做样品是就发现出峰不正常了。本来响应有500的峰高,现在只有50,而且原来不拖尾的,现在尾巴拖的能从3分钟一直到8分钟。我把昨天做过的样品再拿出来分析,也是拖的很严重,根本不成峰了。像是土丘一样。因为跟昨天比,是同样的样品,同样的流动相,(泵的压力也和昨天差不多)同样的波长,不知道为什么会出现拖尾那么严重的,该不会是柱子塌陷了吧,但是我柱子才用20来次,一共也才打了40针。怎么会呢?请遇到或者能解决的大虾帮帮我。[/B][/color][/color]我今天去找修仪器的人来修了,找了半天的原因,最后发现是信号线输出问题。重新插一下,什么问题都没有了!这么简单的问题,哎。我们却一直想的太复杂!!
我们公司的高效液相是安捷伦1200,配备单泵,但是《中国药典》里面的方法是需要双泵梯度洗脱,因此我公司无法按此法检测,现在老板不愿意再买一台泵,要求我从流动相的配比上面来解决单泵洗脱达到双泵梯度洗脱效果的问题,各位专家有什么高招,在此讨论讨论

[font=微软雅黑, sans-serif]【生活中的分析】[/font][font=宋体]毛巾‘脱毛率’,增热度,企业实验室怎么来扩项[/font][font=宋体] 上个月最紧张的纺织品行业事件应该就是毛巾脱毛率事件,甚至一度盖过了口罩的热度,各地工商部门,各地的商场,各大厂商都纷纷自查,看看自己区域有没有曝光的产品,毕竟毛巾产业基地出现问题,全国各地都有牵扯,可忙坏了这个行业的各个环节的人员了。从原材料,到厂家,到商场,到检测单位,到工商人员都动起来了,其实就是因为下面这一句话:‘[/font][font=宋体]这些用再生纤维做成的毛巾“脱毛率没一个达标的”。[/font]’[font=宋体] 作为检验单位,你们嗅到商机了吗?其实就是这个热度过去,毛巾脱毛率也会作为常态重点监控,所以你没有这个项目的检测,你就错过这个大‘蛋糕’了。[/font][font=宋体]想分‘蛋糕’,检测单位该怎么做呢,首先就是找标准,先找毛巾的标准[/font][font=宋体][img=,690,376]https://ng1.17img.cn/bbsfiles/images/2020/08/202008101053114124_2082_2154459_3.png!w690x376.jpg[/img][/font][img=,553,301]https://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif[/img][font=宋体]然后下载标准,打开标准详情查看,找到脱毛率的检测标准[/font][img=,690,387]https://ng1.17img.cn/bbsfiles/images/2020/08/202008101053307575_979_2154459_3.png!w690x387.jpg[/img][font=宋体]继续查阅,下载毛巾脱毛率的方法标准[/font][img=,690,395]https://ng1.17img.cn/bbsfiles/images/2020/08/202008101053455325_6409_2154459_3.png!w690x395.jpg[/img][font=宋体]下载毛巾脱毛率标准后,进行详情查阅,根据标准简介,自己实验室内部制定毛巾脱毛率测试作业指导书。[/font][img=,575,397]https://ng1.17img.cn/bbsfiles/images/2020/08/202008101053557807_3006_2154459_3.png!w575x397.jpg[/img][font=宋体] 然后根据检测项目需求对照自己实验室设备,环境,工具是否具备,最后确立了所需设备实验室都有,具备了该方法对设备和环境检测需求,可以共用设备和检测环境,根据作业指导书进行预运行,进行内部仪器比对,人员培训,人员确认,人员比对。[/font][font=宋体] 并送同一批次毛巾样品到省一级纤检局进行实验室比对检测,得到结果来确定自己实验室检测结果是否满意。[/font][font=宋体]对比结果确认后,然后做方法确认[/font][font=宋体] 因为[/font]GB/T22864-2020[font=宋体]毛巾标准最新标准[/font]2020.9.1[font=宋体]号实施,但是目前的毛巾都是之前生产的,标示都是[/font]GB/T22864-2009[font=宋体]毛巾标准,还是要按照[/font]GB/T22798-2009 [font=宋体]毛巾产品脱毛率测试方法进行检测。[/font][font=宋体] 这个时候扩项[/font]GB/T22798-2009 [font=宋体]毛巾产品脱毛率测试方法是没有必要了,如果要扩项毛巾脱毛率检测,那么按扩项程序要求,进行标准查新后,做方法确认和实验室比对,这期间有单位组织能力验证计划更好,直接扩项[/font]GB/T22798-2019 [font=宋体]毛巾产品脱毛率测试方法,然后赶上[/font]2020.9.1[font=宋体]号标准的实施和运行。[/font][font=宋体] 这次[/font]3.15[font=宋体]晚会毛巾‘脱毛率’为什么被直接爆出来。那是因为旧衣服,内裤,袜子经过机器绞碎再利用,会使得纤维变短,短纤维制成的毛巾,会产生绒丝和毛绒。绒毛极易随呼吸进入鼻腔刺激鼻黏膜,对于有鼻炎患者,会有过敏性反应。就是健康的人也会对呼吸系统有直接伤害。[/font]
抢个第一帖~祝蓝人早日抱个胖娃娃~
今天和一个Q友讨论总磷的测定问题,他总是做的不合格,就逐步分析失败的原因,突然他一句话提醒了我,他说他在900波长的数据不理想,决定用882波长的数据来计算。我问他用的是什么方法,他告诉我是国标啊,水质总磷的测定 钼酸铵分光光度法 GB11893-89 ,而且他还说标准是死的,人是活的,波长这个问题可以做适当地调整,国标上说用700波长,可是测出来的数据不理想,还是这个波长的数据比较理想,我无语了,做化验员好几年,我还真第一次听人这么说。而且他同一项目用不同波长测出一条曲线,认为最大的吸光度对应的波长好,所以全部数据都是在这一波长882测出来的,而且他还认为,不管是什么磷,最终是测那蓝颜色的吸光度,不同的波长下吸光度是不一样的 ,我选的882,只要我的实验全部选用这个波长,就对 。这个问题我觉得我很纠结,原来波长是可以这样活动的,虽然人是活的,标准是死的,但也不能这样把标准搞活吧。原来,标准真的是这样用活的吗?欢迎讨论一下。总磷的国标法GB11893-1989水中的含磷化合物,在过硫酸钾的作用下,转变为正磷酸盐。正磷酸盐在酸性介质中,可同钼酸铵和酒石酸氧锑钾反应,生成磷钼杂多酸。磷钼酸能被抗坏血酸还原,生成深色的磷钼蓝。在700nm波长下,测定样品的吸光度。从用同样方法处理的校准曲线上,查出水样含磷量,计算总磷浓度,用〈P,mg/L〉表示。本法最低检出浓度为0.01P mg/L。
我硅胶柱分离一种二元酸酸,样品可能以钠盐的形式存在,在氯仿,丙酮等有机溶剂中不溶,只在甲醇,水,乙酸中溶解,但是我又害怕样品和甲醇反应生成甲酯,所以不敢用甲醇来冲硅胶柱。我想问能不能把乙酸作为洗脱体系的一种来过硅胶柱呢?
小弟 现在做色素的层析用的是 硅胶 洗脱液是 氯仿/甲醇请问 浸泡硅胶的应该是氯仿 还是甲醇 或是两者的混合物 呢我是湿法上柱的 上样物质就是 粗色素应该用哪种溶剂做吸附剂呢 我糊涂了
想用超高效液相--蒸发光散射监测器测二硝托胺及代谢物,用乙腈:0.1%甲酸水(75:25)等度洗脱时,两者出峰时间基本重合了;梯度洗脱还没弄出来,我该怎样设梯度程序呢 大神指导, 注:代谢物极性大
这个问题可能很菜我的梯度洗脱是水-甲醇-1.2%的乙酸(文献上看的方法)请问甲醇?乙酸?的作用分别是什么??怎么样设计梯度与时间???我现在有的可以跑出来,有的不可以,这是什么原因?怎么样改??
我的前处理条件是0.5克样+5ML硝酸,微波升温20分钟,160度保持20分钟。出来是像汽水一样的蓝色液体。如果按照标准80度加热或者超声脱气2-5分钟的方法,结果明显偏高。后来尝试80度加热30分钟,只有个别标样是在范围,偏高的样还是高的离谱。求助是否有更好的脱气方法或者脱气条件
各位专家:我现在遇到一个问题:我在做水溶性生物碱的盐酸盐分析,样品成分未知,使用agilengt HPLC1100使用99%甲醇和1%水洗脱,结果有三个峰不能够完全分开,流速0.5Ml/min我也试过100%水洗脱结果不理想,我没有加入任何缓冲液和改良剂,因为我打算利用半制备柱进行分离制备,有什么办法使三个峰能达到基线分离呢,而且又不影响我的制备(不引入杂质)?能否通过调节pH来实现,该加何种物质呢?生物碱和其盐在分离时条件一样吗?望各位专家给予解答,谢谢了!
现在我说一下具体情况,我的实验用的仪器是AA6300火陷[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法来做“可溶性”金属总Cr,样品的前处理,取样5克,用0.07mol/l的盐酸在搅拌,并保持PH值在1-1.5间,然后静上过滤,定容到250ml做[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url],我做了四个样品,其中三组样品的含量都是ND,一组吸收值很高,我又稀释了100倍,还是高,然后稀释1000倍,结果测出来是ND,最后是稀释成1000倍测出了计算为103577PPM。因为我是初学者,出于慎重,我把测值很高的那组样品送去SGS检测,结果是报告ND。我看报告上SGS用的仪器是ICP-OES,方法是硝解法。我想,SGS是权威,我可以保证的是我的样品没有受到污染,但我的问题出在哪里呢?是因为使用两种不同的仪器造成了这么大的误差,还是因为处理样品造成的呢?拜托各位指点![em0809]
请问下检测食醋游离矿酸用的百里草酚蓝试纸是什么颜色的?听名字好象应该是蓝色的,可是我做出来的却是黄色的,请各位前辈指教!多谢!![em10]

前言作为纺织品实验室的检测人员,大家不难发现GB /T 22798-2009《毛巾产品脱毛率测试方法》中在很多情况下测试的结果是负数,而作为一种检测脱毛的手段不可能会出现这种结果,下面我就来分析一下测试中的不足…. 一、方法概述将样品放入标准大气中调湿平衡,轻轻抖动以除去落毛,并捡去毛巾上缠绕的落毛和线头,称量产品重量,然后将产品放入洗衣机中按要求洗涤和烘干,再次调湿平衡后称量产品的重量,通过前后两次重量差与洗涤前称量重量的百分比来作为脱毛率的计算结果。、二、试验方法分析1. 负值结果分析:作为毛巾脱毛的衡量标准,一个产品脱掉毛羽后重量不可能增加,只能会减少,所以说出现这种结果一般是由于误差引起的。在该标准的测试中有两次的调湿,而根据GB/T 6529《纺织品的调湿和试验用标准大气》中规定相对湿度的标准范围是65.0±4.0%,也就是说当样品洗涤前调湿的标准大[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]对湿度在下差为61%,而洗涤后样品调湿的标准大[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]对湿度为上差为69%时,中间会有不超过8%的偏差,而产品脱毛的重量比偏差小的话可能会有负差情况;举个例子,干重300g的产品在61%的湿度下重量为300g*(1+0.61)=483g,洗涤后如果脱掉2g的干毛羽,而调湿的环境为69%,则调湿后产品的重量为(300-2)g*(1+0.69)=503.62g,按照标准计算的脱毛率为-4.3%,而实际脱掉的毛羽最小为2g/483g*100%=0.41%,两者一对比结果就很明显了,所以说结果出现负数,很大一部分是由于调湿过程中湿度的变化引起的。[img=,655,177]http://ng1.17img.cn/bbsfiles/images/2017/08/201708201926_01_1954597_3.jpg[/img][img=,562,206]http://ng1.17img.cn/bbsfiles/images/2017/08/201708201926_02_1954597_3.jpg[/img]图1—标准中对温湿度范围的规定2. 计算方式的分析:根据计算方式我们可以知道洗涤前后两者的重量差,不但包括了 脱掉的毛羽,还有一些其他的杂质,因为洗涤过程中洗掉的还有一些染料、灰尘、陪洗织物的脱落毛羽等;[img=,230,157]http://ng1.17img.cn/bbsfiles/images/2017/08/201708201926_03_1954597_3.jpg[/img]图2—洗涤中洗到的染料及杂质三、新法探讨通过对试验方法的分析,我们可以看出方法中的一些不足,针对这种不足,我们是否可以用一种新的方法来代替,下面我就想新方法的可行性做以下分析:1. 新方法概述选择完整的毛巾若干条,室温称重使总重达到1kg,轻轻抖动以除去落毛,并捡去毛巾上缠绕的落毛和线头,然后将所有样品烘干至恒重称量其干重,再按照标准洗涤,洗涤过程中收集洗衣机及排水口中的毛羽,然后将产品进行烘干,再收集过滤网中的毛羽,将洗衣机和烘干机中所收集的毛羽放入烘箱内烘干至恒恒重,计算脱毛率,可用如下公式算得:脱毛率=毛羽干重(洗衣机+排水口+烘箱过滤网)/洗前产品干重*100%2. 方法分析以上方法中通过称量干重,消除了调湿过程中相对湿度的允差影响;收集的毛羽包含了所有洗涤和烘干的损失;3. 设备可行性分析3.1 烘箱称重可参照GB/T 9995方法,采用箱内称重。[img=,381,487]http://ng1.17img.cn/bbsfiles/images/2017/08/201708201927_01_1954597_3.jpg[/img]图3—箱内称重设备外观3.2 洗涤设备毛羽收集:对于有收集毛羽的机型不但可以收集筒内毛羽,还可以收集排水口中的毛羽,排水口下面需要悬挂一个丝网,同时排水口可以安装一个漏斗来缓冲水流。[img=,314,266]http://ng1.17img.cn/bbsfiles/images/2017/08/201708201927_02_1954597_3.jpg[/img]图4- 洗衣机中的收集网[img=,173,190]http://ng1.17img.cn/bbsfiles/images/2017/08/201708201927_03_1954597_3.jpg[/img]图5 - 排水口下面的收集网3.3 烘干设备毛羽收集:一般的烘干设备中都带有收集毛羽的过滤网,这样既可以对烘干过程中的毛羽进行收集测试,还可以减少因毛羽堵塞引起的排风不畅。[img=,189,178]http://ng1.17img.cn/bbsfiles/images/2017/08/201708201927_04_1954597_3.jpg[/img]图6-毛羽收集网[img=,187,113]http://ng1.17img.cn/bbsfiles/images/2017/08/201708201927_05_1954597_3.jpg[/img]图7—毛羽收集网插口总结 通过对以上试验方法的分析判断,我认为收集洗涤和烘干过程中的毛羽应该是衡量毛巾脱毛的最佳方法。也希望大家能给出更加合理的意见。
n 淋洗n 分析物得到保留后,通常需要淋洗固定相以洗掉不需要的样品组分,淋洗溶剂的洗脱强度是略强于或等于上样溶剂。淋洗溶剂必须尽量地弱以洗调尽量多的干扰组分,但不能强到可以洗脱任何一个分析物的程度。n 注意:淋洗时不宜使用太强溶剂,否则会将强保留杂质洗下来。使用太弱溶剂,会使淋洗体积加大。可改为强、弱溶剂混用;但混用或前后使用的溶剂必须互溶 n 洗脱n 淋洗过后,将分析物从固定相上洗脱。溶剂必须进行认真选择,溶剂太强,一些更强保留的不必要组分将被洗出来;溶剂太弱就需要更多的洗脱液来洗出分析物,这样固相萃取柱的浓缩功效就会削弱难道淋洗和洗脱不是一个过程吗
[size=3][/size]我用ASE100提取土壤中的多氯联苯跟有机氯农药,提取剂用的是1:1的丙酮:正己烷,手动过佛罗里希柱,5ml正己烷活化柱子,10ml正己烷:二氯甲烷 1:1的混合液洗脱,回收率才20%多一些,我的过柱的样品只有3ml左右,我不知道该用多少洗脱液来淋洗,请做过的帮忙找下愿意你是不是洗脱的问题。多氯联苯跟有机氯农药的回收率都是这样,旋转蒸发出去的溶剂中没有被测物质,加载样品洗脱前流出的液体中也没有被测物质,初步判断为样品吸附在了柱子上。 我准备做过柱不过柱的对比,不过柱出杂峰但是不对其积分应该也可以吧,做多氯联苯用质谱检测,有杂峰也影响不到。过柱的那部分,分次过柱,我看用多少体积过柱才能使得最后的洗脱液中检测不到被测物质。 有什么好的建议请多多指教
请问各位:我用的是反相色谱,C18的柱子,流动相:A相(1%乙酸 ) B相(纯乙腈),梯度洗脱程序初始比例为A相100% 运行10分钟。再加上之前跑基线平衡大约30分钟也都是用初始梯度。柱子在100% A相的条件下至少运行40分钟。看过资料里说一般不要用低于5%的有机相冲洗柱子过久,否则会引起C18链的卷曲,对柱子造成损害。不知道我天天这样运行对柱子的损害大不大?有什么好的方法调节么?
老师让我测土壤中的氯氰菊酯、莠去津和利谷隆残留量,我想用固相萃取法来净化?(选用的是[B][color=#DC143C]C18[/color][/B]固相萃取小柱),但我不知道选什么淋洗液和洗脱液,只知道要根据极性选,我也看过一些常用溶剂的极性排列表,但还是不知道选什么,因为莠去津和利谷隆是极性较强的,都可以用同一种物质来淋洗和洗脱,但氯氰菊酯是弱极性的,要将这三者同时进行淋洗和洗脱,究竟用什么呢?请大家赶快帮忙出出主意啊,非常感谢!!!
我以前没有做过梯度洗脱的实验,现在我接了一个新实验是这样的流动相C为0.1%TFA,B为0.1%TFA乙腈水(80:20)梯度是这样的:(流速为0.75ml/min)time(min) B% C% 0 0.0 100.030.0 15.0 85.075.0 35.0 65.0115.0 85.0 15.0120.0 100.0 0.0125.0 0 100.0145.0 0 100.0在梯度洗脱之前用什么来平衡柱子(C8),时间从125.0-145.0)这一段时间是不是在冲洗柱子?数据的采集应该是哪一段时间?一般做完实验后用哪一个流动相来冲洗柱子?请大家帮帮忙!谢谢了!
我目前正在做梯度洗脱,但是出来的色谱图惨不忍睹,基线在梯度过程中猛的升高,然后下降,像过山车似的。。。出来的峰是个拱形。。。所以麻烦各位提供一些梯度洗脱的设计中应该注意的地方。谢谢!!
专家你好,我用ESI做保护氨基酸分析时,发现有些保护基团会脱落,我想请教专家,有什么办法能使它,不脱落,我用的是岛津的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]-2010A
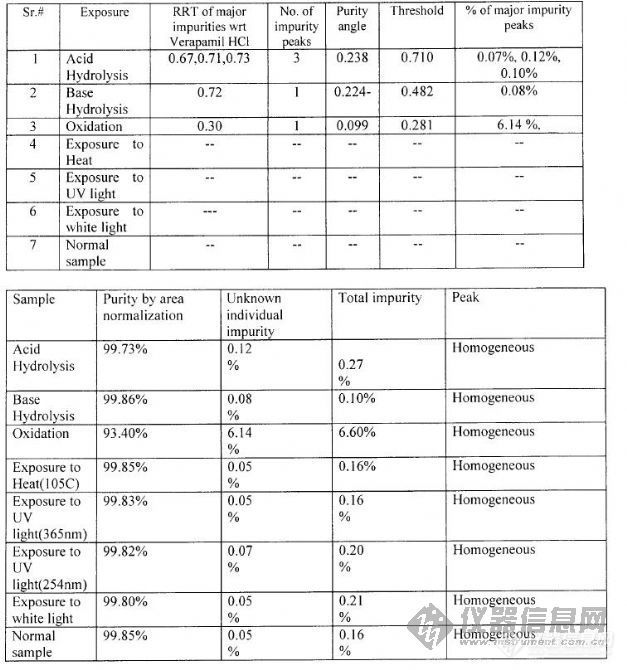
以下是翻译资料时遇到的,以前我一直以为这里的纯度(purity)相当于含量,purity analysis就是assay(含量测定),因为我翻译过很多资料,只有这家公司用HPLC分析过purity,其它公司的资料里没出现过。但现在看起来好像还不是一回事,purity analysis好像是分析杂质的。图见下:有关物质方法学验证中关于强制降解试验的资料:[img]http://ng1.17img.cn/bbsfiles/images/2008/08/200808211341_105135_1612179_3.jpg[/img]我的疑问:1. purity angle(暂且译为纯度角)是怎么计算出来的?怎么会有负值呢(表1第3栏第5列)?这里的负值是像比旋度里的负值一样表示方向呢,还是笔误?2. 从表上看,threshold(暂且译为阈值)貌似是purity angle的阈值,超过它就不合格。那么这个阈值是如何确定的呢?最大是不是不能超过1?3. 表1最后一列中主要杂质峰的百分比,是指峰面积占全部杂质峰面积的百分比吗?4. 表2中的homogeneous(暂且译为“色谱峰均一”)是什么意思?是说色谱峰的峰形好还是指没有共同洗脱的杂质?什么叫共同洗脱呢?5. 为什么要分析色谱峰的纯度呢?看峰形、计算分离度不行吗?6. 在一般的色谱分析中都要分析色谱峰的纯度吗?多谢!
昨天,有一个临时QQ在Q我,我正在想点击查看,突然弹出抖动,一条信息说是求购平焊法兰和无线RTU设备。我一想,不管嘛样先加了再说吧,有业务订单就接啊。加了之后,跟她按正常流程沟通,我说:“您好,有平焊法兰,但是不知道您要什么材质和压力的呢?”A(在此简称A)回答:“碳钢的。”那我又问:“压力等级为多少呢?”A:“1M。”我晕,我说:“您好,压力等级没有1M之说哦,您说的是不是PN1.0Mpa呢?”大家猜怎么着,对方回答我一句话,当场我晕倒在地。A说:“对对,就是这个,人家懒嘛。” 我滴个神啊,我当时更晕了,晕到麻痹的状态倒在沙发上。后来,我又问清楚了这个客户要的具体的尺寸,然后给她报了价格。 唉,我的亲亲客户啊,要是您懒的话,我们怎么报价啊,真是搞不懂啊,如此客户您遇到过吗?提醒,请珍惜帐号,勿发广告~







 我要推广仪器
我要推广仪器
 下载APP
下载APP