[color=#444444]网上查询邻溴苯硫酚沸点:194~196摄氏度,对溴苯硫酚沸点:239摄氏度,色谱柱为Rtx_624,设进样口温度250,检测器270,柱溫160~230的范围,也走过程序升温,分离度最好才是1.0,不知道怎么实验怎么做下去了,请指导一二,另外,准备购买一根极性柱子,思路对不?[/color]
[color=#444444]2,4-二甲基苯硫酚,2,6-二甲基苯硫酚位置异构体。沸点分别为207-208 °C,76℃[/color][color=#444444]使用Scientific TRACE TR-5MS 色谱柱,两个峰出在一起,相差0.1min,无法基线分离。尝试多种升温程序,只有保留时间和峰宽有变化,分离情况不变。降低载气流速也一样。[/color][color=#444444]换用 安捷伦vf-WAXms柱,彻底分不开了。瘦高的一个峰。[/color][color=#444444]两个柱子都试过60,70,90,120度恒温,分不开。梯度升温,也分不开。[/color][color=#444444]请各位大侠帮帮忙,试了两天了。[/color]
邻氨基苯硫酚能不能走液相色谱?有什么要求吗?
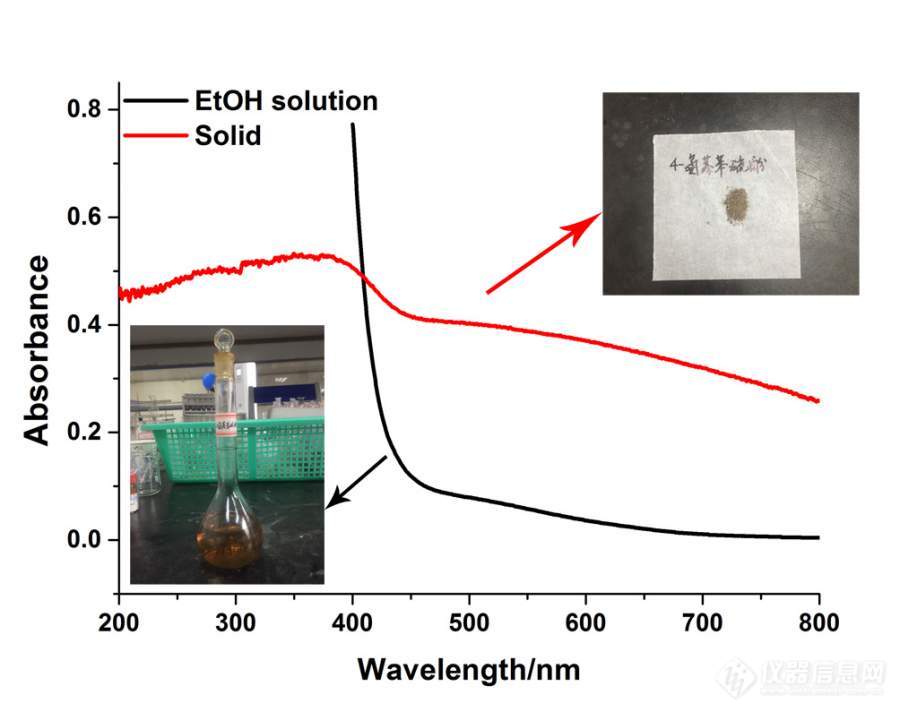
[color=#444444]我最近在做关于对氨基苯硫酚(4-ATP)的实验,我在实验中发现,这个本身是棕褐色的,无论在乙醇溶液还是固体状态,但是它在紫外检测条件下的光谱我发现有些不同。固体条件下,它在可见光区是有着吸收的,但是在乙醇溶液中,可见光的范围内,它的吸收非常低。图中的溶液浓度是10exp(-2)M。我想问问各位,这个应该怎么解释?[/color][color=#444444][/color][color=#444444][img=,690,545]https://ng1.17img.cn/bbsfiles/images/2019/09/201909111034332125_8799_1801607_3.jpg!w690x545.jpg[/img][/color][color=#444444][color=#008000]对氨基苯硫酚(4-ATP).jpg[/color][/color]
1做皮革中的五氯苯酚PCP为什么要用水蒸气蒸馏提取皮革中的五氯苯酚?连内标也要随样品一起进行水蒸气蒸馏。详见标准2而纺织品中的含氯苯酚是用碳酸钾溶液超声提取的3皮革中五氯苯酚水蒸气蒸馏肯定不是模拟,是为了测试总量五氯苯酚,百度了下水蒸气蒸馏,貌似对不溶于水的化合物提取比较有用。4 用其他的有机溶剂如乙醇进行乙醇蒸馏提取应该也可以吧?或者向带液体的样品中直接通N2或压缩空气蒸馏目标物?5 纺织品皮革中的邻苯,PAHS提取理论上也可以采用水蒸气蒸馏提取吧?原理水蒸气蒸馏原理,简言之,就是当水和不(或难)溶于水的化合物一起存在时,整个体系的蒸气压力根据道尔顿分压定律,应为各组分蒸气压力之和。即:P=P水+ PA (PA为与不(或难)溶化合物的蒸气压)当P与外界大气压相等时,混合物就沸腾。这时的温度即为它们的沸点,所以混合物的沸点将比任何一组分的沸点都要低一些。而且在低于100C的温度下随水蒸汽一起蒸馏出来。这样的操作叫水蒸气蒸馏。装置及操作水蒸气蒸馏有两种方法:—种是将水蒸气发生器产生的水蒸气通入盛有被蒸物的烧瓶中,使被蒸物与水一起蒸出;另一种方法是将水加入到装有被蒸物的烧瓶中,与普通蒸馏方法相同,直接加热烧瓶,进行蒸馏,这是一种简化了的水蒸气蒸馏方法;当蒸馏时间较短,不需耗用大量水蒸气时,可采用这种方法。http://ng1.17img.cn/bbsfiles/images/2012/01/201201301051_346869_1689180_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/01/201201301050_346868_1689180_3.jpg
我走的是邻氨基苯硫酚的气质,但是由于原料邻氯硝基苯的量太大,覆盖了产品峰,不知道怎么修改参数让两种物质分离。或者有谁知道能不能用其他办法分离出两种物质
各位老师好,本人写论文需要用到以下化合物的香气或香味阈值数据,麻烦有相关资源的老师帮忙查一下,或者告知如何查找,谢谢!3-巯基-2-丁醇 cas:54812-86-1/ 37887-04-0 四氢噻吩-3-酮 cas:1003-04-9姜烯 姜油酮 cas:122-48-5丁香酚 cas:97-53-0二糠基硫醚 cas:13678-67-62-甲基-3-巯基呋喃 cas:28588-74-1癸醛 cas:112-31-2甲基(2甲基3呋喃基)二硫醚乙酸 cas:64-19-72-甲基苯硫酚 cas:137-06-42-甲基-3-甲硫基吡嗪 cas:2882-20-42-甲基-5-甲硫基吡嗪三乙酸甘油酯 cas:102-76-1柠檬酸三乙酯 cas:77-93-0呋喃酮乙酸酯 cas:4166-20-5甲基糠基二硫醚 cas:57500-00-24-糠硫基-2-戊酮 cas:180031-78-14-甲基-4-糠硫基-2-戊酮 cas:64835-96-7二糠基二硫醚 cas:4437-20-12,4,6-三异丁基-1,3,5-二噻嗪 cas:74595-94-1
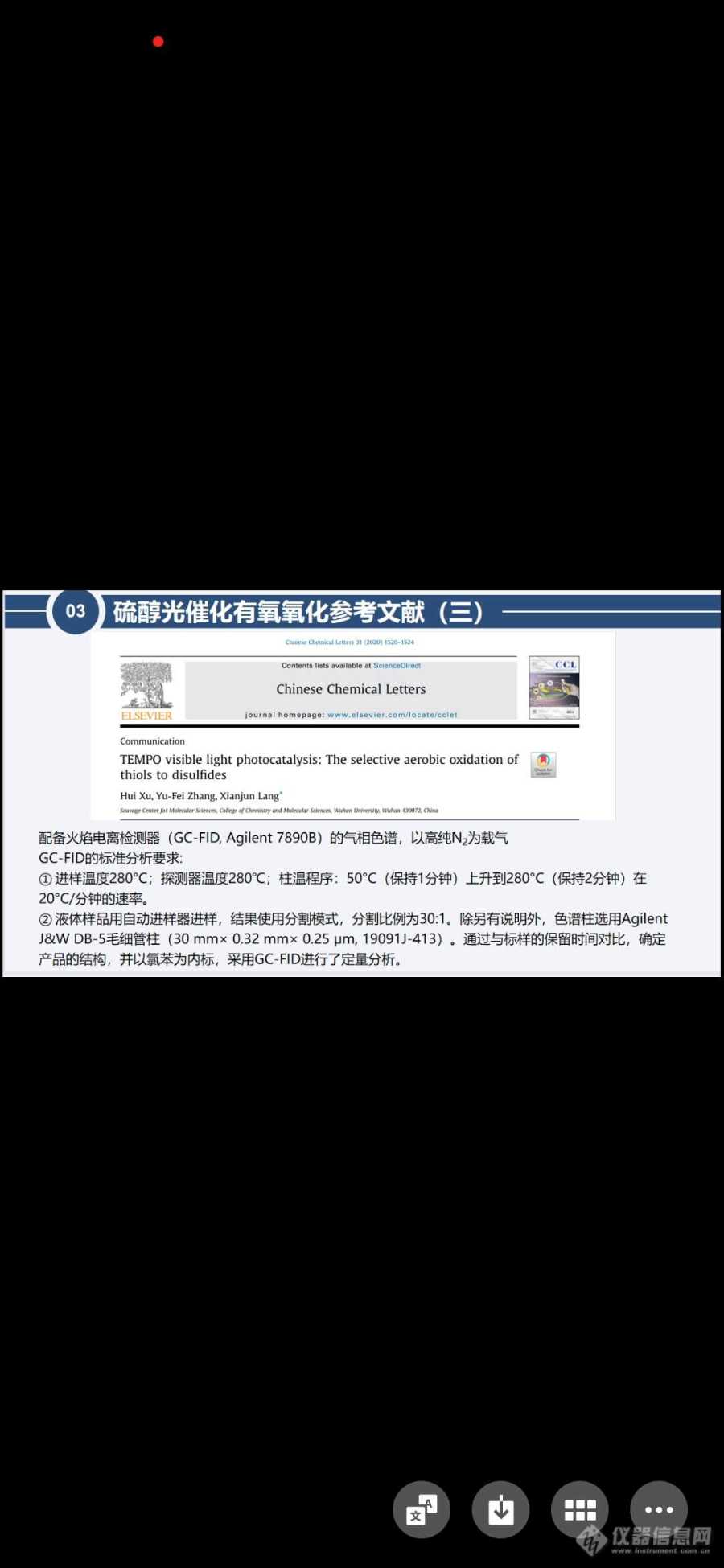
我们用自己的仪器岛津GC2030检测,柱子是用的Rt-5x (30 m 0.25mm 0.25 μm)检测,结果造成色谱柱拖尾了。文献里是用柱子DB-5检测。我想问下能用其他什么型号的柱子@[img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307281006228273_7507_5597722_3.jpg[/img]
空气中苯系物检测标准要求溶剂是二硫化碳,可以用正乙烷代替吗正乙烷能解吸出活性炭管中的苯系物吗?苯酚标液怎么配制方便啊,苯酚结晶太快,不好取啊

SNT 4260-2015 出口植物源食品中粗多糖的测定 苯酚-硫酸法http://ng1.17img.cn/bbsfiles/images/2016/09/201609271937_612349_1779312_3.png
二硫代氨基甲酸酯/盐的多残留测定方法一直是行业难点,分享一篇苯二硫酚衍生测定二硫代氨基甲酸盐类化合物的新方法,为同行提供有益借鉴。
HJ834-2017土壤中半挥发性有机物的测定,硅酸镁小柱对苯酚类化合物有吸附能力吗,还是会直接随溶剂流下去
慕尼黑上海分析生化展,迪马科技最新推出了Inspire 苯基系列色谱柱,一直忙未给大家及时普及,今天来说说第一款Inspire PFP ( 五氟代苯基) 是Inspire 液相色谱柱家族新成员,针对分离极性化合物过程中的保留时间和分离度问题而特别设计。Inspire PFP 凭借其优异的选择性,可为极性化合物、复杂天然产物、位置异构体和其它相关化合物在C18 和C8 色谱柱上的分离提供一个替代和补充。Inspire PFP 具有U 型色谱分离特性,适用于正相、反相和亲水作用色谱三种分离模式,并具有多种作用机理,因而能够同时分离检测不同极性化合物的混合物,为目前难以解决的复杂极性和亲水性样品的分离分析提供了强有力的工具,可轻松解决其它色谱柱面临的分离难题,为用户实现强极性分析物的优异选择性提供一种更加便捷的途径。同时也为色谱工作者使用简单流动相,避免使用极端pH 条件和准备复杂流动相提供了可能性。Inspire PFP 色谱柱特点• 五氟代苯基硅烷键合在高纯硅胶基质上• 具有U 型色谱分离特性,适用于正相、反相和亲水作用色谱三种分离模式• 对极性化合物具有独特的保留能力• 良好的峰形、超高的柱效、分离度和使用寿命• 适用于芳环类化合物或长共轭体系化合物的分离• 优异的批次重现性增强位置异构体分离能力官能团位置的微小差异可以极大的影响分子性能,在许多情况下,传统的C18色谱柱根本无法扑捉到这种细微的差异。然而,Inspire PFP的多功能选择性却可以区分由于分析物内部微小位置变化而导致的分析物的空间位阻变化还是分析物的偶极矩偏移。色谱柱如图所示 规格 150 × 4.6 mm, 5 μm 流动相0.1% 甲酸乙腈溶液:0.1% 甲酸水溶液 = 40:60 流速1.5 mL/min 柱温室温 检测器 UV 254 nm 样品1. 3,4-二甲氧基苯酚 2. 2,6-二甲氧基苯酚3. 3,5-二甲氧基苯酚4. 2,6-二氟苯酚5. 2,4-二氟苯酚6. 2,3-二氟苯酚7. 3,4-二氟苯酚8.3,5-二甲基苯酚9.2,6-二甲基苯酚10.4-氯-3-甲基苯酚11.4-氯-2-甲基苯酚12.3,4-二氯苯酚13.3,5-二氯苯酚http://www.dikma.com.cn/u/image/2016/09/06/1473147613188048.jpg苯氧酸类化合物分子上卤素的加入可以从根本上增强化合物的极性,而极性的变化通常伴随着反相色谱柱在保留时间和分离能力上困难的增加。此时使用InspireTM PFP 是解决保留问题的最有效的方法。InspireTM PFP利用偶极-偶极和氢键作用更好地保留,区分和分离极性卤化化合物。色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相乙腈:0.1% 甲酸水溶液 = 50:50流速1.0 mL/min柱温室温检测器UV 220 nm样品1. 苯氧乙酸2. 邻氯苯氧乙酸3. 对氯苯氧乙酸4. 2,4-二氯苯氧乙酸5. 2,4,5-三氯苯氧乙酸6. 2,4,5-三氯苯氧丙酸http://www.dikma.com.cn/u/image/2016/09/06/1473147817102957.jpg类固醇通过整合偶极-偶极、π-π和氢键机理,InspireTM PFP实现标准反相条件下极性化合物的最佳分离。色谱柱 如图所示 规格 150 × 4.6 mm, 5 μm 流动相甲醇:水 = 60:40 流速1.5 mL/min 柱温室温 检测器UV 254 nm 样品1.泼尼松龙3.地塞米松5.氢化可的松21-乙酸酯7.可的松-21-乙酸酯2.泼尼松4.皮质酮6.11-α羟孕酮8.11-酮孕甾酮http://www.dikma.com.cn/u/image/2016/09/06/1473148006619700.jpg甲基苯乙酮异构体目标分析物上的基团位置变化可以影响化合物的偶极矩,这种变化可以很容易被高电负性的氟原子和其它保留机理察觉,因此InpireTM PFP可以有效地用于分离甲基苯乙酮的位置异构体。色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相甲醇:水 = 50:50流速1.0 mL/min柱温室温检测器UV 254 nm样品1. 邻 -甲基苯乙酮2. 对 -甲基苯乙酮3. 间 -甲基苯乙酮http://www.dikma.com.cn/u/image/2016/09/06/1473148212903667.jpg核苷酸和核苷色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相0.1% 甲酸水溶液流速1.0 mL/min柱温室温检测器UV 220 nm样品1. 胞嘧啶2. 5'-CMP3. 5'-UMP4. 5'-GMP5. 尿苷6. 胸腺嘧啶 http://www.dikma.com.cn/u/image/2016/09/06/1473148405914511.jpg抗胃酸药色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相乙腈:20 mM 磷酸氢二钾(pH 7.0) = 20:80流速1.0 mL/min柱温室温检测器UV 220 nm样品1.法莫替丁2.西咪替丁3.尼扎替丁4.雷尼替丁 http://www.dikma.com.cn/u/image/2016/09/06/1473148597658988.jpg氧化应激标记物色谱柱 如图所示规格 150 × 4.6 mm, 5 μm[/

今天在做检测时,看到标准有一个试剂:新蒸馏的苯酚。不知要如何蒸馏?蒸馏的苯酚与未蒸馏的苯酚有什么区别?感谢“四季风”版友提供的图片及资料:把有结晶的苯酚放置100度C的水浴中溶解,然后倒入蒸馏烧瓶中,放入玻璃珠数粒,瓶口放一支带胶塞200度C的水银温度计,塞紧瓶口(见以下草图)。接好冷凝管,放一接收瓶,在电炉上先加热蒸馏至沸点182度C后约2---3分钟左右,拿去第一个接收瓶,另放一接收瓶接收沸点182度C的苯酚蒸馏液(冷却后即为纯苯酚结晶)。http://ng1.17img.cn/bbsfiles/images/2011/09/201109282355_320046_1641058_3.jpg
做环境空气中苯系物的时候,用二硫化碳解吸,苯的峰分成两半了,有时候不分,有时候分,标液(二硫化碳中苯系物)走出来也是这样,有没有大佬分析下是什么情况[img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108100926585253_264_5343586_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108100926588522_8346_5343586_3.png[/img]
拿到一根新做成的整体柱,如证明他是反相柱 流程如下,找一极性大的苯系物,如苯硫酚, 用甲醇做流动相, 在死时间出峰,随着水相得比例增加,保留时间增加,即证明该柱是反相疏水柱解释如下,,开始时固定相起作用,相似相容,,,反相固定相和样品结合若, 甲醇的洗脱能力强,因此在死时间出峰, 随着水相比例的增加,流动相的洗脱能力减弱,保留时间增加,即证明该柱是反相柱。。。大家探讨正相柱如何证明?
关于五氯苯酚和四氯苯酚的测试。各位买标准物质的时候是买的五氯苯酚和四氯苯酚标准物质,还是直接买已经乙酰化的五氯苯酚乙酸酯和四氯苯酚乙酸酯。买哪种好,为什么呢?
求助SN/T 2145-2008木材防腐剂与防腐处理木材及其制品中五氯苯酚的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法 有此标准的请分享下~~非常谢谢!
[size=5][b]简介[/b][/size] 苯酚-硫酸法是利用多糖在硫酸的作用下先水解成单糖,并迅速脱水生成糖醛衍生物,然后与苯酚生成橙黄色化合物。再以比色法测定。[size=5][b]原理[/b][/size] 多糖在硫酸的作用下先水解成单糖,并迅速脱水生成糖醛衍生物,然后与苯酚生成橙黄色化合物。再以比色法测定。[size=5][b]试剂[/b][/size] 1. 浓硫酸:分析纯,95.5% 2. 80%苯酚:80克苯酚(分析纯重蒸馏试剂)加20克水使之溶解,可置冰箱中避光长期储存。 3. 6%苯酚:临用前以80%苯酚配制。([b]每次测定均需现配[/b]) 4. 标准葡聚糖(Dextran,瑞典Pharmacia),或分析纯葡萄糖。 5. 15%三氯乙酸(15%TCA):15克TCA加85克水使之溶解,可置冰箱中长期储存。 6. 5%三氯乙酸(5%TCA):25克TCA加475克水使之溶解,可置冰箱中长期储存。 7. 6mol/L 氢氧化钠:120克分析纯氢氧化钠溶于500ml水。 8. 6mol/L 盐酸[size=5][b]操作[/b][/size] 1.制作标准曲线:准确称取标准葡聚糖(或葡萄糖)20mg于500ml容量瓶中,加水至刻度,分别吸取0.4、0.6、0.8、1.0、1.2、1.4、1.6及1.8ml,各以蒸馏水补至2.0ml,然后加入6%苯酚1.0ml及浓硫酸5.0ml,摇匀冷却,室温放置20分钟以后于490nm测光密度,以2.0ml水按同样显色操作为空白,横坐标为多糖微克数,纵坐标为光密度值,得标准曲线。 2.样品含量测定: ①取样品1克(湿样)加1ml 15%TCA溶液研磨,再加少许5%TCA溶液研磨,倒上清液于10毫升离心管中,再加少许5%TCA溶液研磨,倒上清液,重复3次。最后一次将残渣一起到入离心管。[b]注意:总的溶液不要超出10毫升。[/b](既不要超出离心管的容量)。 ②离心,转速3000转/分钟,共三次。第一次15分钟,取上清液。后两次各5分钟取上清液到25毫升锥形比色管中。最后滤液保持18毫升左右。([b]测肝胰腺样品时,每次取上清液时应过滤。因为其脂肪含量大容易夹带残渣。[/b]) ③水浴,在向比色管中加入2毫升6mol/L 盐酸之后摇匀,在96℃水浴锅中水浴2小时。 ④定容取样。水浴后,用流水冷却后加入2毫升6mol/L氢氧化钠摇匀。定容至25毫升的容量瓶中。吸取0.2ml的样品液,以蒸馏补至2.0ml,然后加入6%苯酚1.0ml及浓硫酸5.0ml,摇匀冷却室温放置20分钟以后于490nm测光密度。每次测定取双样对照。以标准曲线计算多糖含量。[size=5][b]注意[/b][/size][b][b](1)此法简单、快速、灵敏、重复性好,对每种糖仅制作一条标准曲线,颜色持久。 (2)制作标准线宜用相应的标准多糖,如用葡萄糖,应以校正系数0.9校正μg数。 (3)对杂多糖,分析结果可根据各单糖的组成比及主要组分单糖的标准曲线的校正系数加以校正计算。 (4)测定时根据光密度值确定取样的量。光密度值最好在0.1——0.3之间。比如:小于0.1之下可以考虑取样品时取2克,仍取0.2ml样品液,如大于0.3可以减半取0.1ml的样品液测定。[/b] [/b]
最近要求做水质苯系物能力验证,做过气中苯系物,想请教各位大神,这个水中苯系物曲线用买的标液来配置,溶剂用甲醇还是二硫化碳?用二硫化碳萃取法,需要用水先进行稀释,再进行萃取,该选哪一种柱子比较好呢?本人小白一枚,希望各位大神回复一下,谢谢!
求购测试皮革中五氯苯酚前处理需要用到的高频震荡仪和蒸馏装置,当然装置一定要适合做五氯苯酚的前处理,务必附上图片。我邮箱:cte01@cts-lab.com.cn
最近在看GB5009.19-2008的方法,其中有一个农残 五氯苯基硫醚 从网上找不到CAS号,只能找到甲基五氯苯基硫醚,这两种物质是一样的吗?哪位大侠知道?
大家说说你们测皮革中五氯苯酚是怎么萃取的,是按照标准ISO 17070:2007进行水蒸气蒸馏萃取吗?
如何配制间苯二酚硫酸溶液
想用苯酚硫酸法测多糖,最近看了下但是还有些疑问,求大神指点。1如果苯酚没有变色是不是不用重蒸了,直接配成百分之5的苯酚溶液即可?2苯酚常温下不是固体麽,配制的时候直接称5克溶解定容100ml就可以了不?3一般说来百分之5是现配现用,但也有人说配制好放冰箱保存也可。我想问问,一般配制好的可以存放多久啊?4 加入苯酚后加入硫酸后,摇匀,在90℃的水浴中反应20分钟再在冰水中冷却5分钟。这里硫酸本身就会放热再放到沸水中。。。会不会爆沸啊
整理了几份分析方法,供大家参考!1. 分析对苯二酚葡萄糖苷2. 分析药物中的残留溶剂3. 5类有机磷农药化合物的分析方法
用的科密欧的无苯二硫化碳,为什么仪器老是检测出苯系物。
用苯酚-硫酸法测多糖,在文献里发现不同的苯酚溶液配置方法,有的直接配制,有的要添加Al片、NaHCO3或氢氧化钠后蒸馏。请问苯酚溶液配法不同,对测定有影响么?
各位大侠,有谁做过木材五氯苯酚测试吗?标准要求做五氯苯酚需要用到纯净砂,有谁知道纯净砂是做什么用的吗?在哪里有卖纯净砂的?
我是药残检测的新人,想检测水产品中的五氯苯酚,买不到五氯苯酚标准品,买的是甲醇中五氯酚标准溶液,浓度是100μg/ml。而国标中五氯苯酚标准工作液是用碳酸钾溶液配制,不知如何使用,请教高人指点一下,先谢谢了。







 我要推广仪器
我要推广仪器
 下载APP
下载APP